Cancer Immunology
Hans Schreiber
INTRODUCTION
The goal of this chapter is to provide foundations and key facts in tumor immunology to students, immunologists, and oncologists in order to stimulate critical thinking and experimentation so that we may better prevent, cure, or at least control cancer. The study of cancer has had a tremendous impact on all fields of science (eg, molecular biology, virology, genetics, and immunology). For example, the discovery of the major histocompatibility complex (MHC) by Peter Gorer7,8 clearly separated cancer immunity from autoimmunity. His successful search for specificity led subsequent generations of investigators to search for cancer-specific changes (eg, the consistent chromosome translocation in chronic myelogenous leukemia9). However, conclusive evidence that rejection antigens on cancer cells are cancer-specific came in 1995 when it was shown that they were encoded by somatic cancer-specific mutations.10,11,12 Inherent in understanding the field of cancer immunology is the need to understand that immunology and cancer are two very large, different, and rapidly evolving complex fields of research. Thus, in order to make the immune system effective to prevent or treat cancer, an understanding of cancer and cancer models is required. The discussion of the important roles of cytokines in many aspects of tumor immunology is integrated in the various parts of this chapter.
CANCER
Despite all the progress in molecular biology and genetics, the pathologist using histologic or cytologic criteria13 usually makes the only reliable diagnosis of cancer. Although tumor means “swelling,” the term is usually meant to include cancer cells and the stroma supporting the cancer cells; together they are often referred to as neoplasm, which literally means “new growth.” A neoplasm is an abnormal mass of cells that persists and proliferates after withdrawal of the stimulus that initiated its appearance. Leukemias are cancers caused by neoplastic proliferations of blood cells, but usually do not form tumor masses. There are two types of neoplasms: benign and malignant. The common term for all malignant neoplasms is cancer. Cancers of epithelial tissues (carcinomas) break through the basement membrane to invade adjacent tissues by infiltrative destructive growth. Invasion may or may not be followed by cancer cells entering the lymphatics, bloodstream, or fluid of the coelomic cavities to implant at sites discontinuous with the original tumor (metastasis; Greek for “emigration”). With extremely rare exceptions, metastasis defines a tumor as malignant; benign tumors do not metastasize. Invasion usually precedes metastases and suffices as diagnostic criterion of cancer, although this criterion cannot be used for leukemia and mesenchymal tumors. With the development of inbred mouse strains many decades ago, transplantability of tumors from one syngeneic animal to another became (and still is) a diagnostic criterion for the malignant phenotype of experimental tumors.
Cancer Cells
Many lines of evidence show that cancer is not a single disease. However, there are important principles that apply to many, if not all, cancers. There is substantial evidence that cancers in mice and humans are the result of multiple sequential mutations. As a result, certain molecules in cancer cells are mutant, up- or downregulated, or no longer expressed. In addition, most if not all cancers show epigenetic changes in gene expression. An estimated 15% of the worldwide cancer incidence is attributed to infections,14,15 but chemical and physical carcinogens are involved in the induction of most human cancers in industrialized countries.16 Many of these carcinogens are mutagens.17,18,19 While most mutations in cancer cells seem to be acquired (somatic mutations), increasing numbers of germline mutations are being discovered that make individuals prone to develop cancer.20 A cancer may require as many as 10 or more mutations to develop full malignancy. The first stage of cancer development is called tumor initiation,21 which is generally assumed to be irreversible due to somatic mutations or germline mutations in various oncogene, tumor suppressor gene, or deoxyribonucleic acid (DNA) repair pathways. Initiated cells do not form tumors. However, initiated cells clonally expand to premalignant lesions evolving over many years, often decades. This second protracted stage is driven by tumor promotion (ie, exposure to promoting conditions or chemicals22,23,24 [see Cancer and Inflammation]). The most advanced stage of these premalignant lesions is referred to as intraepithelial neoplasia also carcinoma in situ, often abbreviated as, for example, CIN for cervical intraepithelial neoplasia, VIN for vulvar intraepithelial neoplasia, or PIN for prostatic sites. The premalignant process ends with invasion, the appearance of the first cancer cells. Remarkably, however, there is now clear molecular evidence for premalignant cells spreading to distant sites where these cells remain premalignant unless promoted to become malignant.25,26,27
A cancer cell that survives treatment may cause recurrence of the entire malignancy. Such cells are referred to as cancer stem cells or also tumor-propagating cells.28,29,30 The elimination of these cancer stem cells is critical to prevent relapse. The sweeping assertion that these cells represent an extremely small percentage of the cancer cells in a tumor is not supported by rigorous experimentation or clinical experience. Some of the misconception comes from the observed rarity of only a few human tumor cells able to adapt to growth in a foreign (mouse) milieu.30 Furthermore, flow cytometric analysis of cancer cells may detect a rare population that results from intraclonal and nonheritable heterogeneity in cancer cell populations.31,32
Tumor progression originally meant the change from a benign neoplasm to cancer33,34 but is now usually used to describe the third phase of the multistep process. Invasive growth of a lesion usually ends with a highly aggressive, widely metastatic cancer that ultimately kills the host.35,36 There is compelling evidence that most cancers are clonal in origin and that in cancer progression, new subpopulations of cells arise continuously due to Darwinian selection of genetic variants that have a growth advantage, escape homeostatic controls, or resist destruction by defense mechanisms or treatment.37 During this evolution, sequential mutations result in changes in rate of growth, morphology, hormone dependence, enzyme and cytokine production, and expression of surface antigens. Importantly, by the time cancer is first detected in a patient, it measures usually at least 1 cm in average diameter, contains ˜109 cancer cells, and has already undergone about 30 generations.13 Thus, most of the diversity of a cancer has already occurred at time of detection with only 10 generations left before death of the individual unless treatment intervenes.
The term primary indicates the tumor from which cancer cells emigrate to secondary sites (ie, metastatic growth in tumor-draining [sentinel] lymph nodes or more distant organs). Experimentalists use the term spontaneous metastases to describe metastases that occur without experimental manipulations; artificial metastases are caused by cancer cells injected into systemic or portal veins of a tumorfree mouse to cause lung or liver metastases, respectively. Cancer cells can disseminate without further cell division (microdissemination) or they divide only minimally causing micrometastases, conditions that can only be recognized by microscopy and immunohistochemistry but are potential sources of relapse, the central problem of cancer therapy. Similarly, residual microscopic foci of cancer cells may remain at sites of incompletely excised cancer and cause local recurrence. Efforts are ongoing to develop sensitive markers and assays for determining the need for additional therapy or determining the effectiveness of a therapy before relapse is detected clinically.
Cancer Stroma
Definition
Most of the cells in tumors may not be cancer cells but nonmalignant cells, referred to as stromal cells. Some of the most aggressive cancers, such as pancreatic cancer, mostly consist of nonmalignant stromal cells.38 Virchow39 believed that compression of the growing cancer cells induced a structural fibroblastic framework (now generally referred to as “stroma”) in which the cancer cells grew. He thought that cancer cells and stroma both developed from the same primitive precursors. This concept changed with Ehrlich stating clearly that the host provided the stroma of solid tumors.40 Borst41,42 was the first to clearly point out the essential mutual relationship between cancer cells and tumor stroma by stating that the question of whether the epithelium or the connective tissue has the leading role in carcinogenesis was difficult to answer, because stroma of tumors is dependent on the presence of cancer cells, and cancer cells are dependent on stromal cells. Thus, cancer cells release factors that attract stromal precursor cells, and stromal cells in turn produce factors that support cancer cell growth.43,44,45 Interestingly, Rous46 emphasized the importance of vascularizing stroma for successful tumor transplantation and that immune reaction to nontumor cells led to rejection of the inoculum. Rous, however, was working with noninbred animals. It therefore remained unclear from his experiments whether an immune reaction just to stroma sufficed to cause tumor rejection until 199247 when similar experiments done in inbred mice showed that immune reaction to the stroma of transplanted tumor fragments led to the eradication of the inocula.
Components
Willis, in a careful survey of his own studies and published literature,48 subdivided tumor stroma into just two major components: connective tissue, which usually represents the bulk of stroma, and vasculature, which is usually a smaller fraction. However, we should distinguish at least four critical components: 1) fibroblasts, 2) vasculature, 3) extracellular matrix (ECM), and 4) cells of myeloid and lymphoid lineage, such as macrophages, neutrophils, natural killer (NK) cells, and T and B cells, all derived from hematopoietic stem cells.
Fibroblasts are a prominent cell type in tumor stroma as well as in healing wounds and embryonic connective tissues. Stromal fibroblasts in cancers are metabolically active making matrix substances; the degree of activation of stromal fibroblasts correlates with aggressiveness of the cancer and inversely with survival of patients.49,50 Unfortunately, we still lack reliable fibroblast-specific immunologic markers for these cells despite repeated assertions to the contrary.51,52,53,54,55,56,57,58,59,60,61 Therefore, fibroblasts are still mostly defined by morphology and function.62,63,64,65,66 Characteristically, they synthesize, secrete, and modulate proteins of the fibrous ECM, particularly alpha-collagen.62,63,67 Macrophages are an essential and prominent part of stroma of every tumor. They lack Gr-1, express low levels of MHC class II, and express F4/80. These macrophages are “alternatively” activated (ie, typically have a tumor-promoting immunosuppressive M2 differentiation phenotype, particularly in hypoxic areas of the tumor68). Neutrophil granulocytes, also referred to as neutrophils or polymorphonuclear leukocytes, are an equally pivotal component of tumor stroma. Like the tumor-associated
macrophages, neutrophils in cancers are “alternatively” activated (ie, “N2”69). Unlike classically activated neutrophils, N2 do not cause tissue damage. These neutrophils express cluster of differentiation (CD)11b, Ly6-C, high levels of Gr-1 (defined by monoclonal antibody RB6-8C570), and high levels of Ly6-G (recognized by the monoclonal antibody 1A8).71 RB6-8C5 (anti-Gr-1) recognizes primarily Ly6-G71 but apparently also binds weakly to Ly6-C,71 although this is controversial.72 In any case, RB6-8C5 (anti-Gr-1) not only eliminates neutrophilic and eosinophilic granulocytes effectively (though only transiently),43,44,45,70,73,74 but also markedly decreases cells of the subset of blood monocytes that express high levels of Ly6-C but lack Ly6-G.74 Because of their suppressive effects on cultured T cells,75 these Gr1+ CD11b+ leukocytes are also referred to as myeloid-derived suppressor cells (MDSCs).76,77,78,79 MDSCs are increased in the circulation of tumor-bearing individuals but also found in the stroma of autochthonous80 and transplanted tumors.68
macrophages, neutrophils in cancers are “alternatively” activated (ie, “N2”69). Unlike classically activated neutrophils, N2 do not cause tissue damage. These neutrophils express cluster of differentiation (CD)11b, Ly6-C, high levels of Gr-1 (defined by monoclonal antibody RB6-8C570), and high levels of Ly6-G (recognized by the monoclonal antibody 1A8).71 RB6-8C5 (anti-Gr-1) recognizes primarily Ly6-G71 but apparently also binds weakly to Ly6-C,71 although this is controversial.72 In any case, RB6-8C5 (anti-Gr-1) not only eliminates neutrophilic and eosinophilic granulocytes effectively (though only transiently),43,44,45,70,73,74 but also markedly decreases cells of the subset of blood monocytes that express high levels of Ly6-C but lack Ly6-G.74 Because of their suppressive effects on cultured T cells,75 these Gr1+ CD11b+ leukocytes are also referred to as myeloid-derived suppressor cells (MDSCs).76,77,78,79 MDSCs are increased in the circulation of tumor-bearing individuals but also found in the stroma of autochthonous80 and transplanted tumors.68
Sources
It is presently unknown what percentage of bone marrow-derived stromal fibroblasts originates from hematopoietic versus mesenchymal lineage, what percentage of stromal fibroblasts is derived from circulation rather than local adjacent sources, and whether bone marrow-derived fibroblasts are functionally different.81 As suggested over a century ago, some cells in the stroma come from progenitor cells entering the site of tumor growth either via the blood circulation or from adjacent normal tissues.81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102 The relative contribution of the two sources (circulation or adjacent tissues) is still hotly debated,103,104 but it is questionable whether circulating endothelial precursors contribute significantly to tumor vasculature in well-established tumors. Similarly, it needs to be determined whether local tissue reservoirs are a major source of alternatively activated M2 macrophages in autochthonous or transplanted cancers (> 2 weeks after transplantation) as it appears to be the case in other pathologic conditions of “type 2 inflammation.”105,106 Type 1 inflammation in tumors is an artifact clearly visible during the first 2 weeks after tumor transplantation.68,107 An alternative major source of macrophages in tumor stroma may be monocytes or MDSCs from blood circulation.68
Function
Lack of tumor stroma drastically reduces tumorigenicity.47,108,109 All cancers depend on stromal support and establish some type of paracrine loop.43,44,45,52,110,111,112,113 Signals initiating the loop seem to be intrinsic to the cancer cells (ie, endogenous) and depend on oncogenic mutations in the cancer cells114,115,116,117 (see Cancer and Inflammation). The proinflammatory mediators attract mesenchymal, endothelial, myeloid, and lymphoid progenitors to the stroma from adjacent and systemic reservoirs; the mediators also induce these cells to make factors that stimulate the growth of the cancer cells.43,45,118,119,120,121,122,123,124,125 Cytokines released by transfected cancer cells can have powerful local effects on all components of tumor stroma including fibroblasts.73,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143
Angiogenesis is a fundamental necessity for tumors to grow by allowing oxygenation and nutrients to diffuse from the blood into the mass. Myeloid cells, including mast cells, neutrophils, eosinophils, Gr-1+CD11b+MDSC/monocytes, and tumor-associated macrophages, can promote tumor angiogenesis.144 However, cancer cells and tumor-associated fibroblasts can produce proangiogenic as well as growth stimulatory factors such as vascular endothelial growth factor (VEGF) and Bv8.145 Several studies have shown the importance of neutrophils and granulocyte-colony-stimulating factor (G-CSF) production by cancer cells in causing refractoriness to anti-VEGF therapy.43,44,45,146,147,148,149 Metalloprotease released from neutrophils and from Gr-1+CD11b+MDSC/monocytes catalyzes the release of preexistent VEGF and transforming growth factor (TGF)-β from the ECM and activates latent TGF-β.150,151,152,153 Neutrophils are essential for mobilizing various types of stromal progenitor cells including the macrophages from bone marrow and other reservoirs in the body.154 Finally, neutrophils and Gr-1+CD11b+ monocytes in the tumor stroma themselves can produce large amounts of TGF-β1.69,80
ECM is an essential stromal component providing the cancer cells not only with a scaffold for adherence and structure but also with growth and antiapoptotic signals, thereby preventing anoikis.155,156,157,158,159,160 TGF-β1 stimulates fibroblasts to produce ECM proteins, including collagen, fibronectin, and proteoglycans, and TGF-β1 prevents ECM degradation.161 Accordingly, transfecting cancer cells to produce TGF-β1 makes them more aggressive.128 ECM is also a major reservoir for binding and releasing growth factors, chemokines, and cytokines.162,163 Cancer cells may release the ECM proteoglycan versican that helps attract and activate myeloid cells via toll-like receptors (TLRs) to release interleukin (IL)-6.164 At later stages, neoplasms often replace paracrine with autocrine loops, notably IL-6 activating signal transducer and activator of transcription (STAT)3.123,165,166 However, all evidence suggests that even the most aggressive cancers still depend on some factors and ligands provided by tumor stroma.
Knowing how essential stroma is for cancers to grow, it is somewhat surprising that cancer cells would not generate their own stroma. Indeed, epithelial cancer cells can form a “pseudo-stroma” by assuming a mesenchymal phenotype at the invading edges of the cancer.167,168 However, there is no evidence that this so-called epithelial-to-mesenchymal transition can replace the host-derived stroma (ie, the need of cancer cells to establish paracrine stimulatory loops with nonmalignant stroma169). Fusion of cancer cells with stromal cells, particularly macrophages, has been proposed as a major mechanism of cancer development and progression.170,171,172,173,174,175 However, we still lack conclusive experimental evidence supporting this attractive hypothesis formulated over a century ago.176,177
Reaction to Cancer Cell Inoculation
Careful studies showed decades ago that the many cancer cells that die on inoculation play a critical role in the establishment of the cancer. It was found that adding lethally irradiated cancer cells to an inoculum of viable cancer cells at a 100:1 or larger ratio can increase the take of a cancer cell inoculum by more than a 100-fold.178 The dead cancer
cells have potent thromboplastic activity,179 and death of the cancer cells is required for this activity to form fibrin at the site of inoculation.178 In addition, hypoxia induces VEGF-A and CXCL12 (stroma-derived factor [SDF]-1) in many types of cells, but many cancer cells produce VEGF even in normoxic conditions.180 VEGF-A is a most potent inducer of vascular leakage of plasma proteins, including fibrinogen, that rapidly form a fibrin-fibronectin clot as a provisional tumor stroma.181 Fibrin deposited at the site of inoculation serves as primitive ECM for cancer cells to escape anoikis. The ECM then undergoes major remodeling during the first 2 weeks after inoculation. Remodeling the ECM microenvironment requires the activity of ECM-degrading enzymes such as matrix metalloproteinases.182 Thereby, these transplanted tumors acquire later the harder consistency typical for autochthonous tumors that evolve with stroma.
cells have potent thromboplastic activity,179 and death of the cancer cells is required for this activity to form fibrin at the site of inoculation.178 In addition, hypoxia induces VEGF-A and CXCL12 (stroma-derived factor [SDF]-1) in many types of cells, but many cancer cells produce VEGF even in normoxic conditions.180 VEGF-A is a most potent inducer of vascular leakage of plasma proteins, including fibrinogen, that rapidly form a fibrin-fibronectin clot as a provisional tumor stroma.181 Fibrin deposited at the site of inoculation serves as primitive ECM for cancer cells to escape anoikis. The ECM then undergoes major remodeling during the first 2 weeks after inoculation. Remodeling the ECM microenvironment requires the activity of ECM-degrading enzymes such as matrix metalloproteinases.182 Thereby, these transplanted tumors acquire later the harder consistency typical for autochthonous tumors that evolve with stroma.
EXPERIMENTAL CANCER
Key Principles
Number of Cancer Cells Targeted
A critical determinant for any cancer therapy is the number of the targeted cancer cells that have proliferative potential (“cancer stem cells”). That number determines the likelihood of recurrence/relapse after most cancer cells have been destroyed by treatment. Even microscopic parts of a cancer left behind by a surgeon often lead to recurrence. Nevertheless, the size of tumor-stem cell population is not the only factor determining the likelihood of relapse as other cancer cells, even when dead or lethally irradiated, can increase the tumorigenicity of the remaining cancer cells by orders of magnitude.178
Most human tumors are not detected until they are 0.5 to 1 cm in diameter, have a volume of ˜500 mm3, and contain ˜109 cancer cells.13 In leukemia, malignant cells generally do not form tumors, but the patient has also ˜109 cancer cells when the disease is clinically detected. It is not appropriate to adjust for the difference in host size when comparing cancer in man and mice. Not the size of the species but the size of the cancer cell population determines the chance of relapse because the latter correlates with the number of therapy-resistant variants causing relapse after therapy. Skipper, who pioneered combination chemotherapy of childhood leukemia in the mouse model of L1210 leukemia, targeted 109 cancer stem cells as the starting population. This was one major reason why the principles he established in an animal model were clinically relevant and led to the cure of most childhood leukemias. Cellular heterogeneity within a tumor becomes much more relevant when the tumor accumulates 1 billion (109) cancer cells, equivalent to a tumor with a diameter of 1 cm.183,184,185 At this point, the accumulation of so many cancer cell variants makes it improbable that all cancer cells are susceptible to a single chemotherapeutic agent or specific T cell. Cancer cell variants can be considered analogous to drug-resistant bacteria or viruses (ie, the nature of the problem is fundamentally the same in both cases). Unfortunately, in most experimental studies on immunotherapy, almost fivefold smaller populations of cancer cells are being treated4 (Fig. 47.1).
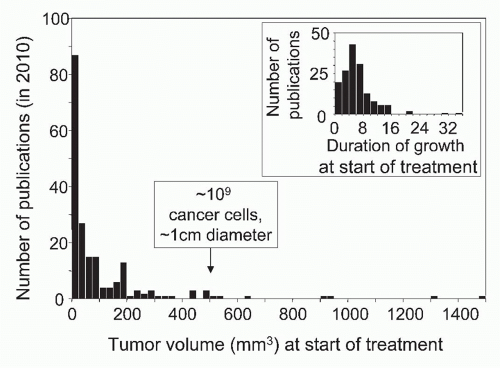 FIG. 47.1. Precipitous Drop in the Number of Publications Reporting the Effects of Experimental Cancer Immunotherapy with Increasing Size or Duration of Growth of Tumors Treated by Immunotherapy. A search of PubMed for year 2010 publications using the keywords “immunotherapy” AND “cancer” recovered 195 experimental studies listing tumor volume or size and 158 studies listing duration of growth at start of treatment. Seventy-five percent of tumors treated in these studies were smaller than 121 mm3. Only 9 of the 158 tumors had grown for 2 weeks or longer. Modified from Wen et al.4 |
Duration of Growth
Duration of growth of a cancer greatly influences experimental results. Summarizing three decades of studies on immunity to cancer, Woglom concluded in 1929 that immunotherapy is futile against an established tumor, and “nothing may accordingly be hoped for at present in respect to a successful therapy from this direction.”186 Eighty-two years later, the outlook is not quite so grim, but unfortunately, the vast majority of research in animal models is still concentrating at treating malignant cell populations grown for an average of only 5 days after cancer cell inoculation4 (see Fig. 47.1). The word “established” is not a scientific term, yet is frequently used to describe neoplastic lesions caused by recently implanted cancer cells,186 conveying the message that the malignancy being treated does not differ from what would be found in a human cancer patient. Just the opposite is true; transplanted tumors must grow for at least 2 weeks before they are histologically indistinguishable from autochthonous murine or human cancers.107 Most human cancers have resided in the patient for months if not years before being detected and treated, whether primary, metastatic, dormant, or relapsing. An additional problem of experimental models is that growth of many serially transplanted “standard” cancer lines is so rapid that death may occur so early that treatment has to be started before solid tumors have established a microenvironment even vaguely comparable to that of an autochthonous tumor.
Measuring Growth and Destruction
Tumors are masses and have weight and volumes best approximated by the formula of an ellipsoid (V = πwh/6 or V = 0.5236 lwh or V-½ lwh); length 1, width w, and height h are the orthogonal diameters in the three perpendicular
axes. As was carefully documented, the third dimension, height (or depth), has an inordinately large effect on errors in volume187 but can usually be determined accurately for subcutaneous tumors.187,188,189 Describing tumors as areas is incorrect and illogical despite widely practiced misuse. By definition, areas have no volume nor contain a single cancer cell. Area measurements as commonly used by radiologists frequently disagree with volumetric measurements.187,188,190 Depth of a tumor can greatly vary and determines malignancy and prognosis (eg, in melanoma).
axes. As was carefully documented, the third dimension, height (or depth), has an inordinately large effect on errors in volume187 but can usually be determined accurately for subcutaneous tumors.187,188,189 Describing tumors as areas is incorrect and illogical despite widely practiced misuse. By definition, areas have no volume nor contain a single cancer cell. Area measurements as commonly used by radiologists frequently disagree with volumetric measurements.187,188,190 Depth of a tumor can greatly vary and determines malignancy and prognosis (eg, in melanoma).
There are important variables when extrapolating the number of cancer cells from a given tumor volume; for example, the human or murine adenocarcinoma of the pancreas usually consists predominantly (˜90%) of nonmalignant stromal cells yet is highly aggressive, whereas in most tumors stromal and cancer cells are more balanced in numbers. Furthermore, while it takes weeks for a tumor to disappear completely even after all cancer cells have been eradicated, it is usually impossible to determine from volume measurements how many viable cancer cells are left. Certain cancers release hormones that have systemic effects such as insulin; even microscopic growth can have major systemic effects.191 While many experimental models utilize subcutaneous tumors, some cancers, particularly human tumor xenografts, may require transplantation to sites where it is more difficult to assess growth objectively.192 Cancer cells have been transduced with luciferase to give a signal proportional to the tumor size when substrate is injected; this allows whole body scans with a luminometer, though sensitivity decreases for fewer than 1,000 cancer cells, and there is absorption by overlying tissues.193
Cure and Dormancy
The effects of the immune system on cancer can be read out in different ways. Certainly, cure of the cancer is the objective. There should be clear prolongation of survival and absence of relapse, but proving that all cancer cells have been eradicated is often difficult. Unfortunately, for most cancers, we lack assays sensitive enough to detect and quantify remaining dormant cancer cells. This information would indicate whether or not a patient requires further treatment. Only in a few types of cancers194,195,196 does a negative diagnostic polymerase chain reaction analysis of the tissue reservoir from where a cancer may relapse suggest—though not prove— complete eradication. Experimental evidence for dormant cancer cells may come from provoking relapse by treating the animal with antibodies neutralizing factors or cells suspected of causing tumor dormancy. Experimentation should include waiting for minimally 30 days after the tumor has disappeared completely (it is best to wait several months or longer). A common unacceptable practice is using the word eradication or cure when there is no follow-up after the cancer becomes undetectable. Relapse of cancers may occur within days, weeks, months, or even years after complete disappearance, and is one of the most important problems of cancer therapy. Nevertheless, experimentalists commonly describe treatments as effective even when followed by rapid relapse.197 Eradication means tearing a tree out with its roots so it cannot regrow, and the term is synonymous with cure. Thus, using the term eradication is only appropriate if the host does not harbor dormant cancer cells, comparable to “sterilizing immunity” in infections.198,199,200,201
Inhibition, Arrest, Regression, and Equilibrium
Dependent on the extent and type of destruction as well as when it occurred, tumors may be found to have smaller or larger volumes after treatment compared to controls. It is important to determine whether the rate of tumor growth has been altered or the rate of growth remained unaltered but the onset of growth has changed. Altered rates of growth require an ongoing process, whereas altered onset of outgrowth is usually caused by an event that happened at times of inoculation. Slower growth rates (growth inhibition) should be distinguished from shrinkage (regression) of a cancer. A steady size of a treated tumor compared to controls is referred to as growth arrest, equilibrium, or progression-free survival in cancer patients, an important goal when cure cannot be achieved.169,202
Specificity Controls
It is completely inappropriate yet quite customary to study immunologic therapies specific for a self-antigen on a human cancer in mouse models (human tumor xenograft) when the murine host does not express the same target. These irrelevant models often yield impressive results and mislead the reader that the targeted molecule is tumor-specific when in the real situation it is not.203,204 If someone subscribes to the widespread highly questionable perception that many, or even most, useful tumor antigens are self-antigens, then this investigator should also use appropriate experimentation. Either a mouse model must be used in which normal tissues express the target molecules closely resembling expression patterns in man or there should be a clear warning to the reader. For example, anti-carcinoembryonic antigen (CEA) immune responses caused toxicity in mice that expressed the target antigen also in normal tissues, as humans do,205 predicting the severe toxicity later observed in a clinical study.206 Side effects often become apparent only when the treatment is intense enough to provide clinical efficacy.207 It is therefore questionable to dismiss results in proper animal models.208,209
Selection of Tumor Model
General Considerations
Experimentalists have to be able to translate their findings to clinicians and vice versa. There is no single human cancer, let alone single animal model, that can serve as appropriate model for all human cancers.28 Organ site as well as histologic type of a cancer may greatly influence the results. Thus the complexity of cancer makes it extremely important that the experimental model used to study cancer immunity be relevant to the question asked; a single model, if carefully chosen, may be appropriate to answer a specific question. Nevertheless, it is essential that we uncover the broader principles underlying cancer-host interactions. For example, the extensive clinical and experimental research on immunotherapy of melanoma has failed to answer the
central question: why are we struggling to make similar advances in the immunotherapy of more common cancers such as breast, colon, prostate, and lung cancers? Weiss argued in 1980 that the failures of clinical immunotherapy were due to using irrelevant laboratory tumor models for extrapolating results for clinical application.210 One of the major reasons for translating results from animal models to clinical cancer immunotherapy being frustratingly evasive is the disregard of using truly established tumors for experimental therapy (see Fig. 47.1).4 Current experimentation in cancer immunology mostly uses young hosts to study the effects of the immune system on small, recently implanted inocula of cancer cells derived from tumors once induced in inbred mice (with homozygous genetic loci) and then passed in vivo for decades from animal to animal, whereas human cancers are already well established when first detected, have a very high probability of heterozygous loci, have never been transplanted, and develop in mostly older individuals.
central question: why are we struggling to make similar advances in the immunotherapy of more common cancers such as breast, colon, prostate, and lung cancers? Weiss argued in 1980 that the failures of clinical immunotherapy were due to using irrelevant laboratory tumor models for extrapolating results for clinical application.210 One of the major reasons for translating results from animal models to clinical cancer immunotherapy being frustratingly evasive is the disregard of using truly established tumors for experimental therapy (see Fig. 47.1).4 Current experimentation in cancer immunology mostly uses young hosts to study the effects of the immune system on small, recently implanted inocula of cancer cells derived from tumors once induced in inbred mice (with homozygous genetic loci) and then passed in vivo for decades from animal to animal, whereas human cancers are already well established when first detected, have a very high probability of heterozygous loci, have never been transplanted, and develop in mostly older individuals.
Modes of Induction
Spontaneous. The term spontaneous cancer is defined as cancers arising “in the absence of any experimental manipulation.”211 Spontaneous murine cancers develop without any known exposure to carcinogens or genetic (often transgenic) manipulations introducing oncogenes or eliminating tumor suppressor genes. For example, spontaneous tumors can be observed in many mouse strains with advancing age. All genes have a spontaneous rate of mutation, and genetic instability, though often a consequence, may not be a requirement for tumor development.212,213 As would be expected from the sporadic occurrence of spontaneous mutations, the occurrence of spontaneous tumors is random and unpredictable.
Carcinogen-Induced (Physical, Chemical, or Viral). Many of the physical and chemical carcinogens involved in the induction of cancers are mutagens.16,17 Since repeated application of coal tar was used to induce the first chemically induced cancers,214 a very large number of chemicals such as polycyclic hydrocarbons and nitroso compounds has been identified with remarkably potent cancer-inducing activity.215 Cancers have been induced in many tissues and organs and several animal species. Ultraviolet light (UV) has potent skin cancer-inducing activity in man and mice,216,217 and many well-defined models of UV-induced tumors are now available.218,219 The potential of ionizing radiation to cause cancer in humans was recognized soon after Roentgen’s discovery of x-rays in 1895.220,221 But study of radiation carcinogenesis in animals mostly occurred after World War II when large-scale tumor-induction studies were carried out in many species over the succeeding three decades in response to the threat of irradiation from nuclear reactors or bombs.221,222,223 Finally, many models of viral cancer induction have been developed after Rous224 showed that viruses can also induce cancer in animals.
Transgene-Induced. Experimental cancers are produced artificially by inserting oncogenes into the germline of mice or by manipulating the mouse genome to allow excision of tumor suppressor genes. Genes used for these manipulations are driven by tissue- or cell lineage-specific promoters that are either constitutively active or inducible locally or systemically. While these mice become prone to develop cancer, the tumors they develop are autochthonous but by no means “spontaneous” despite widespread misuse of terminology.
Autochthonous Tumors
Autochthonous tumors originate in the place where they were found (autochthonous, in Greek, means indigenous). The antonym is transplanted tumors. Autochthonous tumors can be spontaneous, carcinogen-induced, or transgeneinduced. Even when induced in the same experiment by the same mode of induction, autochthonous tumors will differ genetically, biologically, and antigenically from one another, because additional but individually differing genetic changes are required for each tumor to develop. Thus autochthonous tumors lack the uniformity of well-defined transplantable models. An advantage of autochthonous over transplanted tumors, however, is that the host’s immune system has been neither artificially primed nor altered by an inoculum.
Unlike autochthonous tumors developing after exposure to physical, chemical, or viral carcinogens, transgeneinduced autochthonous tumors have the disadvantage that transgene expression in the thymus during development usually causes systemic tolerance to the transgenic proteins. Nevertheless, several excellent transgenic cancer models have been developed.225,226 Some models are based on immunologic findings first made in patients.12 For example, the R24C mutation in the cyclin-dependent kinase 4, first identified as tumor-specific antigen by T cells of a melanoma patient, causes familial melanoma when in the human germline.227 Introducing this mutation in the germline makes mice highly susceptible to develop melanoma225; tumor development, however, requires additional carcinogenic insults followed by prolonged chemical promotion.
Certain oncogenes such as SV40T are powerful because they inactivate several important suppressor pathways and may therefore require fewer additional mutations. In some transgenic cancer models, the transforming genetic event can be controlled, sometimes reversibly, by topical or systemic application of an inducer such as tamoxifen or tetracycline.193 Alternatively, systemic or topical application of Cre-recombinase may excise a “floxed” blocking element or attenuator of gene expression.228 These temporal controls appear to be helpful for answering important questions on tolerance, because expression at birth causes neonatal tolerance to a highly antigenic oncogene such as SV40 T antigen. An approach that more closely mimics the sporadic nature of human cancer relies on a spontaneous mutational event activating an introduced floxed oncogene. The sporadic nature of this event unfortunately also makes time and site of tumor development less predictable.228
Transplanted Tumors
Most current experimental work in tumor immunology uses transplantable tumors. However, there are substantial differences among transplantable tumors, and it is
important that the chosen cancer is appropriate for the particular question being asked. It is important to know that 24 hours after inoculation, the majority of the injected cancer cells usually have died,178 leaving only a shallow outer rim of viable cancer cells at the oxygenated margin.107 Histology or in vivo imaging reveals pronounced edema at the injection site. In fact, much of the “tumor growth” early after inoculation is due to tissue swelling caused by the inflammatory reaction. Cancer cells may well be invading surrounding normal tissues and be vascularized within 48 to 72 hours after transfer, thereby meeting standard criteria for malignancy. But experienced pathologists will immediately spot the abnormal inflammatory reaction to the transplantation injury and the necrotic center of the early inoculum. Thus, common terms such as “three-day established cancer” or “three-day established metastases” are highly misleading. Such necrosis would not be found in an autochthonous cancer of similar size. The inflammatory reaction progressively decreases with time, and at 14 days, the necrotic material has usually disappeared. The tumor then contains ˜109 cancer cells, measures ˜1 cm diameter, and is histologically indistinguishable from primary autochthonous tumors. Ideally, therapy uses tumors 2 weeks or more after inoculation (see Fig. 47.1).
important that the chosen cancer is appropriate for the particular question being asked. It is important to know that 24 hours after inoculation, the majority of the injected cancer cells usually have died,178 leaving only a shallow outer rim of viable cancer cells at the oxygenated margin.107 Histology or in vivo imaging reveals pronounced edema at the injection site. In fact, much of the “tumor growth” early after inoculation is due to tissue swelling caused by the inflammatory reaction. Cancer cells may well be invading surrounding normal tissues and be vascularized within 48 to 72 hours after transfer, thereby meeting standard criteria for malignancy. But experienced pathologists will immediately spot the abnormal inflammatory reaction to the transplantation injury and the necrotic center of the early inoculum. Thus, common terms such as “three-day established cancer” or “three-day established metastases” are highly misleading. Such necrosis would not be found in an autochthonous cancer of similar size. The inflammatory reaction progressively decreases with time, and at 14 days, the necrotic material has usually disappeared. The tumor then contains ˜109 cancer cells, measures ˜1 cm diameter, and is histologically indistinguishable from primary autochthonous tumors. Ideally, therapy uses tumors 2 weeks or more after inoculation (see Fig. 47.1).
Even though the terms tumor transplantation and tumor injection are old and commonly used, they are quite misleading, as they usually refer to inoculation of tumor fragments or a suspension of cancer cells. A common error is the belief that tumors can be injected or “transplanted” like a vascularized organ such as kidney or heart. Instead, much of the inoculum—whether a cancer cell suspension or fragments of a tumor—dies initially and needs about 2 weeks before it becomes histologically indistinguishable for autochthonous tumors.
Tumors can be induced in animals by injecting cancer cell suspensions prepared from cultures or by mincing tumors into 1-mm3 pieces and injecting the tumor fragments using a 12-gauge trocar. Cancer cells in fragments are 10- to 100-fold more tumorigenic than stroma-free cancer cell suspensions.47,108 It had long been known that certain cancers would only grow in mice when transplanted as tumor fragments. This was erroneously thought to be due to more cancer cells being inoculated with fragments.229 Later analyses revealed that fragments contained fewer cancer cells than injected cell suspensions, yet produced a higher take or larger tumors earlier.47,108 Another erroneous explanation was that the stroma of tumor fragments provided a physical barrier preventing cancer cells from migrating to draining lymph nodes and priming a protective T-cell response.108,230 More likely, more cancer cells remain viable when embedded in tumor stroma (by preventing anoikis160) and therefore release less antigen than suspended cancer cells, most of which die. In any case, as long as cancer cells express sufficient levels of antigen, professional antigen-presenting cells in the tumor stroma pick up the antigen and travel to the draining lymph nodes where they present the antigen to naïve T cells.231
It is important to know that whether suspensions of cancer cells or tumor fragments are being used, a threshold number of cells or fragments must be inoculated for tumors to develop. The threshold in T cell-deficient mice may be due to innate immunity or nonimmunologic mechanisms.178,232 The increase in threshold in immunocompetent mice is probably due to adaptive immunity.
Cell Lines from Tumors Serially Passed in Mice. When choosing tumors that have been serially passed in animals for transplantation, it is critical to be aware of how such tumors were altered by the serial transplantation. As was discovered decades ago, even a single in vivo passage of a cancer can select for heritable cancer variants.45,219,233,234,235,236,237 Even half a century ago, investigators already noted that serial transplantations of these cancers “inevitably result in progression toward more rapid growth rate, loss of functional and histological differentiation, loss of responsiveness to extraneous stimuli”238(p.522) and a diminution of strain specificity, a problem shared by B16.239 These serially transplanted cancers, such as B16, can easily be transferred in mice using cancer cell suspensions rather than tumor fragments. As a result of hundreds of passages in mice,239 these cancer cells have become resistant to anoikis caused by lack of stroma by acquiring alternate signaling pathways that replace the prosurvival signals of ECM.240 Many of these tumors grow and kill so fast that they must be treated early before they are truly established. Certainly, many of these tumors no longer resemble primary mouse or human tumors that virtually always grow at much slower rates and have been established for months or years before being treated.
B16 melanoma was derived in C57BL/6J mice in 1954 and is the parent of many available B16 sublines.239 B16 had been transplanted serially through 328 mice for 13 years before it became available as a standard cancer cell line. Nevertheless, B16 still is arguably the “Escherichia coli” of tumor immunology with almost 1,000 entries in PubMed in 2011. The A/J-derived Sa1 originated in 1947 in an A albino mouse and was serially transplanted in A/J mice for 1,017 generations for 19 years. Similarly, the A/J-derived neuroblastoma 1300 derived in 1940 was serially transplanted for decades from mouse to mouse. The Lewis lung carcinoma, isolated by Lewis,241 arose spontaneously in 1951 at the Wistar Institute in a black C57 mouse (not a C57BL/6 mouse), was serially transplanted extensively, and is still being used in numerous studies as a model for exploring the immune responses to lung cancers and their metastases. When genetically inbred mouse strains became available as sources for murine cancers over half a century ago, serial transplantation was necessary for maintaining a tumor. Dependable long-term cryopreservation did not become available until the late 1960s and early 1970s.242,243,244 Even after dependable cryopreservation became available, investigators still continued to use serial transplantations to propagate newly derived cancers such as the BALB/c-derived CT26 colon cancer,245 the C57BL/6-derived MC38 colon cancer,246 and the BALB/cCr-derived RENCA renal cancer.247 Many cell lines are renamed sublines of old parental cell lines. Thus, it is often overlooked that RMA, RMA-S, MBL-2, and EL-4 tumors are derived from the same single tumor line,248 most likely
E.L.4 induced by 9:10-dimethylbenzanthracene in 1945 by Gorer249 and serially transplanted for many decades from mouse to mouse. Importantly, referring to EL-4 as a C57BL/6 tumor is incorrect as this strain did not exist in 1945. Similarly, Neuro2a is a subline derived in 1969 from the A/J-derived neuroblastoma 1300 derived in 1940. Unfortunately, organizations providing these lines and/or publications often fail to cite the fact of long-term in vivo passage before cryopreservation.
E.L.4 induced by 9:10-dimethylbenzanthracene in 1945 by Gorer249 and serially transplanted for many decades from mouse to mouse. Importantly, referring to EL-4 as a C57BL/6 tumor is incorrect as this strain did not exist in 1945. Similarly, Neuro2a is a subline derived in 1969 from the A/J-derived neuroblastoma 1300 derived in 1940. Unfortunately, organizations providing these lines and/or publications often fail to cite the fact of long-term in vivo passage before cryopreservation.
Cell Lines from Autochthonous Mouse Tumors. Autochthonous cancers never need to be serially transplanted. Standard lines can be created by adapting the autochthonous cancer cells to culture and/or by freezing fragments or cells in liquid nitrogen.218 To prove that antigens expressed by the malignant cancer are truly tumor-specific, nonmalignant cells, tissue and DNA from the host of tumor origin must be available.218,219 This approach is commonly used by researchers studying the antigenicity and genetics of human cancers. Carefully controlled experimental cancers exist.218,219
Human Tumor Xenografts. For certain purposes, human tumor xenografts are useful models. Human cancer cell lines grown in vitro can cause tumors when injected into T-, B-, and/or NK-deficient mice. It is important to remember that the stroma of such cancers is entirely of mouse origin and the cancer cell-stromal loops are defective because of multiple mismatches in ligand-receptor signaling.250 This problem can be partially alleviated by “humanizing” the recipient mouse. Humanizing usually refers to expressing certain human molecules such as receptors/cytokines in mice that lack murine T, B, and/or NK cells, and/or transferring human mesenchymal and/or hematopoietic cells to such mice. Interestingly, human T- and B-lymphocytes and fibroblasts contained in human tumor fragments may coengraft thereby “humanizing” the mouse192 (eg, when injecting cell clusters or aggregates from human fresh ovarian cancers into nonobese diabetic-severe combined immunodeficiency IL2γR null mice). Progressive growth of these xenografts leads with great regularity to ascites formation, and pleural metastasis closely simulating classical tumor progression observed in patients with ovarian cancer.192 Despite continuous improvement, mice can never be completely humanized. Only very few cancer cells of the xenografts may be able to progress in the chimeric milieu.30 Nevertheless, once a tumor grows, its sensitivity to potential therapeutic agents might reveal the sensitivity of the original cancer growing in the patient.
Selection of Recipient/Host
The vast majority of human cancers develop in later midlife and old age,251,252,253,254 and there is clear evidence that, at comparable ages, mice have difficulties rejecting immunogenic cancers.255,256,257,258 Yet most experimental studies use young mice. Mice should have a clinically relevant tumor burden or be selected for treatment when the bulk of cancer cells has been removed to undetectable levels by surgery or chemotherapy, but dormant cancer cells stay behind to cause later relapse. T cell-deficient mice have been used extensively as models for adoptive transfer of T cells.259 However, cancer patients are usually T cell competent and capable of generating regulatory T (Treg) cells that may not allow an effective “take” of the transferred T cells unless the recipient is lymphodepleted.
CANCER ANTIGENS
No term in cancer immunology is more important and confusing than the term cancer antigen (or tumor antigen). Any molecule detected with T cells or antibody on the surface or within cancer cells is commonly referred to as cancer antigen. The usefulness of a cancer antigen for detection and destruction depends on its specificity (ie, that the antigen is not common to normal cells). The first well-defined tumor antigens encoded by the MHC were discovered by Gorer.8 Immunity to MHC antigens will kill the cancer cells but also the host because of the ubiquitous expression of MHC on normal cells.
General Aspects
Early History
In the late 1800s,260,261 it was discovered that in some instances tumors developing spontaneously in experimental animals could be transplanted into other animals of the same species and in this way could be propagated continuously. This finding provided an important experimental tool for cancer research.40,262 Immediately, scientists began to investigate the possibility of immunizing against such transplantable cancers. Rodents exposed to a small nonlethal challenge of certain tumors became immune to subsequent challenge with large transplants of the same tumor that regularly killed nonimmunized recipients. Also, complete removal of the transplanted tumors, after initial growth, immunized animals against that tumor. These early results seemed to suggest that immunization against cancer was possible. Furthermore, there were certain other spontaneous tumors that were not readily transplantable, and this was taken as evidence for “natural resistance” or “natural immunity” to the cancers. Many years later, it became clear that no such conclusions could be drawn from these early studies, because outbred, or incompletely inbred, rats or mice had been used. The problem became apparent when it was realized that the immunization with tumor would also immunize the host against normal tissue of the donor and that normal tissues of the donor could also immunize the host against the tumor.186 These experiments brought the idea of tumorspecific antigens into disrepute but also started the search for antigens that caused rejection of normal transplanted tissue. This research eventually led to the discovery of the MHC and to the development of inbred mouse strains.8,263,264 Once inbred mouse strains became available, it was found that cancers transplanted within an inbred mouse strain usually grew so well that the existence of tumor-specific antigens seemed very unlikely. In fact, transplantability of tumors in syngeneic animals became (and still is) a diagnostic criterion for the malignant phenotype of an experimental tumor. This criterion was especially useful because many rodent
tumors are cancers of nonepithelial origin (sarcomas) and also because a clear histologic demonstration of local invasive growth can be especially difficult in such cancers.
tumors are cancers of nonepithelial origin (sarcomas) and also because a clear histologic demonstration of local invasive growth can be especially difficult in such cancers.
Proof of Specificity
After discovery of the MHC and the development of inbred mouse strains, the gloom over tumor immunology ended with the discovery that inbred mice could be immunized against syngeneic sarcomas induced by the chemical carcinogen methylcholanthrene (MCA). The first demonstration of induced immunity to a transplantable MCA-induced sarcoma was by Gross in 1943265; however, it was not until the 1950s that more complete experiments provided unequivocal evidence for “tumor-specific” rejection of transplanted cancers.229,266,267,268,269,270 In particular, the experiments of Prehn and Main268 in 1957 made it likely that the rejection antigens on the MCA-induced sarcomas were functionally tumor-specific, because transplantation assays could not detect these antigens in normal tissues of the mice used. However, irrefutable evidence for the existence of tumorspecific antigens in autochthonous unmanipulated cancers only came after a 50-year-long search3,5,218,219,236,267,268,269,270,271,272,273,274,275,276,277,278,279,280,281,282 when in 1995, it was proven that tumor-specific antigens on cancer cells were encoded by somatic cancer-specific mutations absent in the normal cells of the host of tumor origin.10,11,12
Even though it is clear that cancer-specific antigens are encoded by somatic cancer-specific mutations, the terms “tumor-specific” or “cancer-specific” are frequently used inappropriately, sometimes even used in conjunction as “relatively” tumor-specific. The discussion is far from being semantic. In reality, an antigen either is or is not tumor-specific. Germline controls are absolutely critical for proving mutations are somatic and tumor-specific.283,284,285,286 Unfortunately, germline controls are missing from virtually all tumors used for experimental work today. Nevertheless, mouse tumors with proper autologous controls have also been used and are available for distribution.218,219 Notorious problems are genetic polymorphisms. For example, even after 20 backcross generations when a strain is arbitrarily pronounced “inbred” (because it is then more than 99.9% genetically identical), about 373 polymorphic proteinencoding loci remain allogeneic.287 Any one of these loci could encode a pseudotumor-specific antigen when a tumor is transplanted into a mouse misperceived to be fully syngeneic. Mice respond preferentially to nonself- or mutant-self-antigens whether caused by genetic polymorphism or tumor-specific somatic mutation.
Peptide Antigens and Major Histocompatibility Complex Affinity
T cell-mediated destruction of cancer cells requires the interaction of T-cell receptor (TCR), peptide, and MHC molecules. In this “three body problem,” two affinities simultaneously determine the interaction288,289: the peptide to the MHC and that of the TCR to the peptide-MHC complex. Even when the complex cell-cell interaction is reduced to the three molecules interacting in vitro, biochemical analysis is still too complex for analyzing physiologic interactions. TCR affinity to peptide-MHC is therefore usually measured by plasmon resonance in the presence of saturating nonphysiologic amounts of the peptide, and TCR affinities measured this way range between (Kd) 1 to 100 µM.288,289 This is a narrow range considering that affinities of peptides to the MHC range from 1 to more than 20,000 nM.290 This difference points at the peptide-MHC affinity probably being the greatest variable and emphasizes the paramount importance of choosing target peptides with highest possible affinity to the presenting MHC. Proper selection is particularly important as the amount of peptide produced by the cancer cell may be relatively small and always must compete for binding with all other peptides naturally present in the cancer or in the cells cross-presenting the antigen. Affinity of a peptide to a given MHC molecule is best measured empirically and many empirical affinities are already available (eg, http://tools.immuneepitope.org or www.syfpeithi.de).290,291,292 However, even when a peptide binds with high affinity to MHC, it will only be expressed on the cell surface when it is naturally processed and present in sufficient amount.293,294
Antigens Revealed by “Reverse Immunology”
“Reverse immunology” is the attempt to predict T-cell epitopes within a given amino acid sequence. Traditionally, “reverse immunology” has focused on finding optimal T-cell antigens, properly referred to as peptide epitopes, on infectious agents for generating vaccines. More recently, self- or mutant proteins recognized by T cells or antibodies from cancer patients have been analyzed in the search for peptide epitopes that may be effective targets of T-cell immunity. Computerized algorithms (eg, http://tools.immuneepitope.org295,296 and www.syfpeithi.de297) have been developed to predict the affinity of peptide-MHC binding, appropriate proteasomal cleavage, and transport by the transporter associated with antigen processing (TAP).290,291,292,297 Though these algorithms are useful, it appears that these tools cannot replace empirical biochemical measurements. Many of the antigens proposed by reverse immunology have affinities to MHC insufficient to serve as effective targets when tested in appropriate animal models. Together, high peptide-MHC affinity is essential but not sufficient to predict that a given peptide serves as an efficient target.
Complete sequencings of cancer cell genomes from individual patients is becoming increasingly affordable and has revealed up to many thousands of mutations per cancer cell.298,299,300 However < 1% of these mutations cause amino acid substitutions. For example, of the 33,345 nucleic acid base substitutions found in a human melanoma, most of the mutations were intergenic, intronic, noncoding, silent, or truncating. Only 187 caused amino acid substitutions in coding genes,300 and only a few of these 187 coding substitutions are expected to lead to mutant peptides that serve as effective targets. To be effective, the mutant peptide 1) must bind with a high affinity (IC50 in the nM range) to the particular MHC molecules of that individual and 2) be naturally processed, 3) escape destruction by proteasome cleavage, and 4) be present in sufficient amounts. Only a few of the myriad of tumor-specific mutations identified in human or murine cancers give rise to antigens that fulfill these requirements. However, the frequency of unique tumor-specific targets in
a cancer cell may be larger than predicted when considering only mutations in coding sequences, because mutations in intron sequences can be translated and also encode tumorspecific antigens.11 These mutations are over 50-fold more frequent than those in protein-encoding loci.
a cancer cell may be larger than predicted when considering only mutations in coding sequences, because mutations in intron sequences can be translated and also encode tumorspecific antigens.11 These mutations are over 50-fold more frequent than those in protein-encoding loci.
Antigens Recognized by Patients’ T cells
In the search for the best antigens to eradicate the cancer, T cells infiltrating tumors (TILs) have been recovered, expanded, and used therapeutically. However, TILs are removed from cancers that have not been destroyed. It is, therefore, a widespread misperception that the TILs were necessarily fighting cancer growth. Three questions become obvious: 1) Are strong antigens retained because the host has been tolerized during very early stage of cancer growth? 2) Can T cells specific for the strongest antigens be recovered from the TILs? 3) Can T cells specific for weak antigens promote rather than inhibit tumor growth?
It is possible that 1) potent rejection antigens are retained because they tolerize the host very early during cancer development,228,301 2) a few T cells have not been tolerized and can be propagated for therapy, and 3) tolerization is reversible, and competent effector cells can be obtained from TILs. Clinically, the patients who fare best have T cells in their peripheral blood that are specific for antigens encoded by somatic tumor-specific mutations.12,302,303 Whether the same T cells were infiltrating these cancers as TILs is unknown. In any case, it is likely that only a few (if any) of an endless array of antigens recognized on cancer cells by antibodies or tumor-infiltrating T cells may have significance as targets or diagnostic markers.
Cancer-Specific Antigens (Encoded by Mutant Genes)
Prevalence
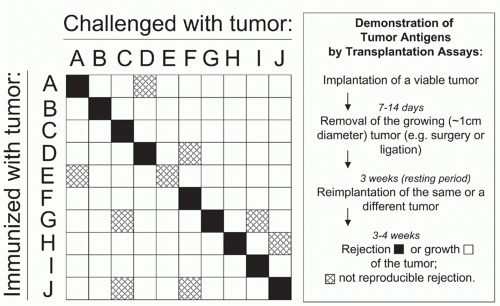
All cancers in man and mouse that have been analyzed carefully express bona fide tumor-specific antigens that could be targeted by T cells. Because tumor-specific antigens arise from mutations,10,11,12 they are usually unique. Each patient’s cancer seems to have a unique set of mutations, and unique antigens can provoke powerful immune responses. In mice, immunization with one tumor protects only against the same tumor (Fig. 47.2).5,274 Shared cancer-specific antigens do exist when the same mutations occur in several cancers, but by comparison, such antigens are relatively rare. Most cancer-specific mutations affect intracellular proteins that may be recognized by T cells as mutant peptide-MHC complex on the surface of viable cancer cells. Very few cancer-specific mutations affect surface proteins, such as the mutant epidermal growth factor receptor (EGFR), a shared tumor-specific antigen on glioma cells.304,305
Oncogenicity
A major misconception is that unique cancer-specific antigens are caused by random mutations and are incidental to the oncogenic process. For example, a group of experts
stated in 2009306(p.5326) that unique antigens come “from random mutations presumed to be present in all patients,” implying mutations encoding unique tumor antigens have no functional significance in the malignant process. Nothing is further from the truth. Of course, all mutations caused by chemical and physical carcinogens are random, but virtually all of them are lost during cancer evolution except those that promote the neoplastic process. When it was first shown in 1995 that cancers harbor cancer-specific antigens caused by somatic tumor-specific mutations,10,11,12 every one of the three antigens represented a mutation in a tumor suppressor locus.307,308,309,310,311 For example, the mutation in the cycline-dependent kinase 4 reduces the binding to its inhibitor and tumor-suppressor protein p16INK4a,12 and the same mutation is found in the germline in cases of familial melanoma.227 The fact that selection for a tumorspecific antigen commonly represents a mutation in genes with functional significance in the malignancy of that cancer (oncogenes or tumor suppressor genes) has been shown in many other studies.219,312,313,314,315 Remarkably, T cells against unique tumor antigens identify mostly novel, functionally important tumor-specific mutations and oncogenic proteins that would not have been easily detected by other technologies.10,11,12,219,307,308,309,316 Importantly, several of these unique tumor-specific antigens are excellent targets because they cannot be lost by immune selection. The reason is that some of these mutant proteins are not only oncogenic but are also needed to provide an essential household function no longer provided by the second allele due to Knudson-type loss317 or mutational inactivation.302,307,308,309
stated in 2009306(p.5326) that unique antigens come “from random mutations presumed to be present in all patients,” implying mutations encoding unique tumor antigens have no functional significance in the malignant process. Nothing is further from the truth. Of course, all mutations caused by chemical and physical carcinogens are random, but virtually all of them are lost during cancer evolution except those that promote the neoplastic process. When it was first shown in 1995 that cancers harbor cancer-specific antigens caused by somatic tumor-specific mutations,10,11,12 every one of the three antigens represented a mutation in a tumor suppressor locus.307,308,309,310,311 For example, the mutation in the cycline-dependent kinase 4 reduces the binding to its inhibitor and tumor-suppressor protein p16INK4a,12 and the same mutation is found in the germline in cases of familial melanoma.227 The fact that selection for a tumorspecific antigen commonly represents a mutation in genes with functional significance in the malignancy of that cancer (oncogenes or tumor suppressor genes) has been shown in many other studies.219,312,313,314,315 Remarkably, T cells against unique tumor antigens identify mostly novel, functionally important tumor-specific mutations and oncogenic proteins that would not have been easily detected by other technologies.10,11,12,219,307,308,309,316 Importantly, several of these unique tumor-specific antigens are excellent targets because they cannot be lost by immune selection. The reason is that some of these mutant proteins are not only oncogenic but are also needed to provide an essential household function no longer provided by the second allele due to Knudson-type loss317 or mutational inactivation.302,307,308,309
Therapeutic Significance
Another misperception surrounding unique tumor-specific antigens is that they have remained unexploited clinically, because truly personalized therapy would be required. Again, nothing is further from the truth. One of the most effective immunologic treatments of cancer today is the adoptive cell transfer of autologous tumor-infiltrating lymphocytes from patients with metastatic melanoma: response rates are in the range of 50% to 70% of the patients, and a few of these patients are cured.318 This truly personalized therapy involves reinfusion of the patients’ own lymphocytes isolated from the patients’ own melanomas and expanded in vitro. Because the TILs response is dominated by T cells to unique tumor-specific antigens,303 it is likely that the success of the reinfused T cells depends on their reactivity to unique tumor-specific antigens. Similarly, clinical studies show that therapeutic vaccinations with autologous cancer cells are likely to be much more effective,319,320 confirming decades of experimental work.229,234,268,269,271,321,322 Self-antigens may serve as useful and effective targets in a very few instances (eg, CD20 and CD19), but industry supported by government panels and organizations focuses almost exclusively on self-antigens,306,323,324 so far with very modest results.325 Finally, it is a misperception that immunizing each patient to the unique antigen of the individual’s tumor is impractical because of cost and should not be pursued. This is incorrect compared with other highly individualized treatments such as those used in renal transplantation. Thus, individualizing cancer therapy could be affordable as other strategies used in the clinics.
Shared Tumor-Specific Antigens
Ideally, antigens targeted on cancers would be expressed exclusively on malignant cells but be shared by cancers of the same type or at least subtype. Several cancer-specific mutations have been identified that are shared between cancers,9,304,326,327 but very few of them have so far been found to be an effective immunologic target. The main reason probably is that, aside from specificity, additional requirements for effective recognition by T cells are 1) high-affinity binding to the patients’ MHC molecules and 2) expression of the protein at sufficient amounts to compete with other peptide for presentation. Unique tumor-specific antigens must fulfill the same requirements but, because these unique mutations are more abundant, chances are much higher to yield an effective antigen. A few examples of shared tumor-specific antigens are discussed in the following.
Mutant Epidermal Growth Factor Receptor. See further under Immunotherapy.
Fusion Proteins. Fusion proteins found in cancer cells are the result of internal deletions (see mutant receptor EGFRvIII under Immunotherapy) or chromosomal translocations.9,326 New antigenic determinants can result from the juxtaposition of previously distant amino acid sequences, resulting in a mutant peptide sequence at the breakpoint and possibly a change in conformational structure. The same chromosomal breakpoints consistently recur in different individuals with the same cancer, therefore result in shared tumor-specific antigens. Fusion proteins encoded by these translocations are usually essential for cancer maintenance, making them ideal targets for pharmacologic and immunologic intervention because tumor cells may not easily escape therapy by losing expression of the fusion proteins.328 Pharmacologic approaches are exemplified by the drug imatinib targeted to the fusion protein of the 9;22 translocation in chronic myelogenous leukemia.9,294,329 The antikinase drug imatinib does not selectively inhibit the catalytic activity of the BCRABL fusion protein, and this results in toxicity to normal cells in the patient. The intracellular BCR/ABL fusion proteins can be recognized specifically by antibody but only in fixed cells.330 BCR/ABL fusion peptides can also be recognized by human CD4+ T cells in the context of MHC class II331 and serve as targets for CD8+ human cytolytic T cells.332 Why do we then still lack effective immunologic therapies for targeting this and other fusion proteins? As explained previously, the predicted epitopes may 1) not be generated naturally by cells,293 2) lack sufficient affinity to the particular MHC of that patient, or 3) not be produced in sufficient amounts.
Mutant RAS. Point mutations in oncogenes can also be shared by several cancers and could encode useful antigens. For example, a valine for glycine substitution at position 12 of RAS is one of the most common mutations in human cancers and can be recognized by human CD4+ T cells.333 The region of the mutant RAS protein from which
the peptide was derived is identical for all the three members of the RAS proto-oncogene family, namely, H-RAS, K-RAS, and N-RAS, which have different prevalence in different cancers. In addition, about 90% of pancreatic adenocarcinomas, a very aggressive human cancer, have one of three to four different single amino acid substitutions in codon 12 of the cellular Kirsten RAS gene.334 Despite remarkable clinical and experimental efforts,335,336,337,338,339,340 targeting mutant RAS-derived epitopes has so far met with little if any success, the likely reasons being the same as those mentioned under the section titled Fusion Proteins. Immunizing cancer-prone mice harboring “initiated” mutant ras-expressing cells with mutant ras peptide resulted in mutant ras-specific CD4+ T cells, antibodies to the mutant ras protein, and more tumors and faster tumor development.341 Possibly, the antibody, produced in response to antigen, activated myeloid cells via FcR-γ to become tumor-promoting, and/or antibody carrying TGF-β downregulated CD8+ effector responses.342,343
the peptide was derived is identical for all the three members of the RAS proto-oncogene family, namely, H-RAS, K-RAS, and N-RAS, which have different prevalence in different cancers. In addition, about 90% of pancreatic adenocarcinomas, a very aggressive human cancer, have one of three to four different single amino acid substitutions in codon 12 of the cellular Kirsten RAS gene.334 Despite remarkable clinical and experimental efforts,335,336,337,338,339,340 targeting mutant RAS-derived epitopes has so far met with little if any success, the likely reasons being the same as those mentioned under the section titled Fusion Proteins. Immunizing cancer-prone mice harboring “initiated” mutant ras-expressing cells with mutant ras peptide resulted in mutant ras-specific CD4+ T cells, antibodies to the mutant ras protein, and more tumors and faster tumor development.341 Possibly, the antibody, produced in response to antigen, activated myeloid cells via FcR-γ to become tumor-promoting, and/or antibody carrying TGF-β downregulated CD8+ effector responses.342,343
Self-Antigens (Encoded by Normal Genes)
All antigens listed in this section are encoded by nonmutant cellular genes and expressed not only by cancer but also by at least some normal adult cells. Therefore, these antigens are not tumor-specific and are commonly referred to as tumor-associated antigens. The level of expression of these antigens can vary from widespread expression to restriction to a small population or a subset of normal cells. However, self-antigens should not be called “quasi-tumor-specific” because even very low levels and/or selective expression of a self-antigen may cause severe even lethal toxicity when targeted.204,206
Self-antigens expressed by tumor cells are used for destruction, inhibition, or detection of cancerous growth. When used as targets for destruction, T cells or antibodies must eliminate the cancer cells while not destroying normal cells expressing the same self-antigen, or destruction of selfantigen -expressing cells must be tolerated. Thus, no serious toxicity must occur even when the immunity is strong enough to destroy the cancer cells.195,196 Antibodies against growth factors or their receptors can inhibit growth of cancer cells without destroying them. When self-antigens are used for diagnosis, background levels of antigen generated by normal cells complicate use of these antigens for early detection of cancer. However, changes in the amount of circulating self-antigens may indicate relapse of cancer after therapy.
All normal individuals have so-called natural autoantibodies as well as T cells to a wide spectrum of self-antigens without causing pathology, but those B-cell receptors and TCRs have usually very low affinity. In fact, any immune receptor binds with some affinity to any particular antigen344 and immune receptors may bind to several molecularly unrelated structures,345 making the discussion of specificity seemingly useless. However, the antibody response of an individual to self- and nonself-antigens differs in affinity by many orders of magnitude, and therapeutically effective antiself-antibodies or TCRs are commonly raised in a nonself, usually xenogeneic, setting. Whether removing natural mechanisms that prevent autoimmunity is a general approach to achieve truly effective antitumor immunity needs to be substantiated.
The problem of B-cell unresponsiveness can be overcome by immunizing mice with xenogeneic human antigen. Misleadingly, these high-affinity antixenogeneic antibodies are usually advertised as “fully human” when they were made in mice in which the murine immunoglobulin (Ig) gene locus had been inactivated and replaced with the human Ig gene locus.346 The toxicities of such “fully human” antibody will still be those of high-affinity antiself antibody: severe to lethal toxicity. These reactions may occur when the variable region of such antibodies (eg, anti-CEA or anti-human epidermal growth factor receptor [HER]-2204,206) is fused with the transmembrane and signaling domains and then transduced as chimeric antibody receptors (CARs) into T cells.347
T-cell tolerance to self can be overcome by making T cells in a host that is allogeneic to the MHC class I molecule presenting the targeted peptide.348 However, recent studies exposed that such allo-human leukocyte antigen (HLA)-restricted high-affinity T cells can have severe “off-target” reactivity likely to cause toxicity when used in patients.349 T cells transduced with these TCRs kill each other (fratricide) when the self-antigen (eg, wild-type p53 or survivin) is expressed by the T cells.350,351 High-affinity TCR cells can also be generated in knockout mice lacking the targeted selfpeptide (eg, wild-type p53 peptide sequences352), but T cells expressing these TCRs are lethal when given to normal mice because wild-type p53 is expressed in bone marrow and can also be expressed by most other normal cells.350
Overexpressed Molecules
Growth Factors and Their Receptors. See Immunotherapy.
Survivin. Although survivin is overexpressed in many cancer cells, it is a problematic antigen particularly as a vaccine because it is widely expressed on lymphocytes causing fratricide.351 Not surprisingly, this antigen has been ineffective in vaccine trials.
p53. Research using p53 as target for immunotherapy is exemplary of the problems and persistent misconceptions of research on tumor-specific (unique or shared) and selfantigens. Mutations in the p53 suppressor gene are among the most common found in human and experimental cancers.353,354 These mutations tend to cluster in evolutionarily conserved regions of the gene, but the exact locations are highly diverse in individual cancers.355 Targeting these mutations would require an individualized therapy considered by many impractical. Therefore, researchers have attempted to exploit the fact that mutant p53 is usually overexpressed in cancers. Thus, major efforts have been made to target conserved, nonmutated regions of the p53 protein hoping for “relative tumor specificity” based on the usual belittlement for low-level expression of this protein by every normal cell including lymphocytes. As a result, T cells expressing highaffinity anti-p53 TCRs commit fratricide unless the anti-p53 TCRs are transduced into T cells from p53 knockout mice.
However adoptive transfer of these T cells causes lethal hematopoietic ablation in normal mice.350 Mortality can be prevented by reconstitution of the recipients with bone marrow from p53 knockout mice,350 an option unavailable in humans. Not surprisingly, there is no evidence for clinical efficacy of targeting wild-type p53 in humans.
However adoptive transfer of these T cells causes lethal hematopoietic ablation in normal mice.350 Mortality can be prevented by reconstitution of the recipients with bone marrow from p53 knockout mice,350 an option unavailable in humans. Not surprisingly, there is no evidence for clinical efficacy of targeting wild-type p53 in humans.
Prostate-Specific Membrane Antigen. Prostate-specific membrane antigen (PSMA) is a type II transmembrane glycoprotein more correctly referred to as glutamate carboxypeptidase II. PSMA is an inappropriate misnomer because it is anything but specific for prostate; it is also expressed by duodenal mucosal cells, proximal renal tubule cells, and a subpopulation of neuroendocrine cells in colonic crypts,356 and has important functions in the brain.357 Vaccination of mice with human PSMA induces antitumor effects without causing toxicity but because the mouse model lacks expression of human PSMA on normal tissues, the findings in this inappropriate model are meaningless.358 Similarly, a “fully humanized” anti-PSMA antibody had potent antitumor xenograft activity in mice that lacked expression of this antigen on normal cells.359 Therefore, this antibody may have severe, possibly lethal, toxicity in patients.
Differentiation/Lineage-Specific Molecules
Some antigens expressed on tumor cells are also expressed during at least some stage of differentiation on nonmalignant cells of the lineage from which the tumor developed. Differentiation antigens may therefore help to determine the organ or cell type of origin (lineage) of a cancer.360 For example, B-cell tumors express surface Ig, and T-cell leukemias can be separated into helper and suppressor cell leukemias using T-cell subset-specific monoclonal antibodies to surface and intracellular antigens including transcription factors. For metastatic cancer of unknown origin, differentiation antigens can be important indicators of the histologic and organ site of the primary tumor.361 Careful diagnostic delineation of different subtypes of cancer is important because different tumor subtypes may have different prognoses and may be susceptible to different therapies. However, the use of differentiation markers for histologic or cytologic tumor classification has pitfalls.361 Lineage-specific antigens represent a very diverse group of proteins: glycoproteins (including mucins) and glycolipids (carbohydrate, peptide, glycopeptide, or glycolipid epitopes). Several of these antigens are being explored as potential immunotherapeutic targets. However, the normal cell types that express these antigens will determine toxicity and usefulness of any antibody or agent for which these are targets.
Cluster of Differentiation 20 and Cluster of Differentiation 19. CD20 is a signature B-cell differentiation antigen targeted by a genetically engineered monoclonal antibody that is relatively effective in the treatment of B-cell lymphoma. Lineage restriction of this marker limits the cytocidal effects to long-term depletion of normal B cells. This depletion is usually well tolerated because patients can be protected by intravenous administration of IgG. While antibodies to CD20 antibodies are therapeutically effective, relapse is common. CD19, but not CD20, appears to be expressed on the more immature malignant cells causing the relapse. Therefore, anti-CD19 Fv has been used in the killing of these cancer cells, either as a fusion protein with an anti-CD3 Fv to engage T cells195 or as chimeric antibody receptor inserted into the patient’s T cells.196 These treatments may prevent relapse.
Melanocyte-Specific Differentiation Antigens. Several differentiation antigens (such as tyrosinase, the related brown locus protein or tyrosinase-related protein 1 (Trp-1), gp100, and Melan A/MART-1362,363) appear to be restricted to melanocytes, and all of them are being explored as immunotherapeutic targets in melanoma.364,365,366 Immune recognition of the melanocyte differentiation antigens can lead to rejection of a tumor challenge but this self-antigen-specific immunotherapy not only targets the tumor cells but also normal cells expressing the shared antigens,365,367 resulting in the depigmentation of normal skin (vitiligo) and possibly other, more serious toxicities (see following discussion). For example, mice immunized with syngeneic Trp-1 in various adjuvant settings fail to produce a T- or B-cell response.368 Xenogeneic or altered Trp-1, however, induces responses that cause vitiligo and protect against challenge with melanoma cells.368 Passive transfer of Trp-1-specific antibodies from these mice causes vitiligo and protects against metastatic spread of melanoma cells in mice when given at the time of seeding of the malignant cells.367 Similar antibodies may have analogous beneficial effects in human melanoma patients,369 but evidence is lacking for such antibodies eliminating bulky human melanoma. Trp-1-deficient mice respond to Trp-1 as a foreign antigen and Trp-1-specific CD4+ T cells from these mice can eradicate large B16 melanoma 10 to 14 days after inoculation,370,371 cause autoimmune vitiligo, and damage the retina.370 Designating these T cells as “tumor-specific” when they are clearly not is incorrect and misleading.370 Trp-1 is expressed in all neurocrest-derived pigmented cells, not only those of the skin but also those of the eye (uvea), inner ear, and brain (substantia nigra, forebrain, and midbrain).372,373,374,375 Other targets include gangliosides GD2 and GD3 that are also not only overexpressed in melanoma but are also found in other cells of neurocrest origin and in other tissues.376,377
Prostate-Specific Antigen. Prostate-specific antigen (PSA), also called kallikrein-3, is a chymotrypsin-like protease that digests semenogelin I and II to release motile sperm.378 Elevation of PSA above the normal range occurs in inflammation (prostatitis) and benign hypertrophy of the prostate as well as in prostate cancer. Using PSA levels for early detection of prostate cancer is controversial. The U.S. Preventive Services Task Force no longer recommends this test in healthy man.379 However, detection of any PSA following complete surgical removal of the prostate indicates residual tumor cells and/or recurrence.380
Prostate-specific phosphatase is selectively expressed and secreted by the epithelial cells of the prostate gland.378,381 Sipuleucel-T, a vaccine targeting prostate-specific phosphatase
and approved by the U.S. Food and Drug Administration for the treatment of castration-resistant prostate cancer, results in only a very modest improvement in overall survival.323 Furthermore, a recent analysis of internal documents that became available after the U.S. Food and Drug Administration approval questions the adequacy of the trial and correctness of the conclusions drawn.382
and approved by the U.S. Food and Drug Administration for the treatment of castration-resistant prostate cancer, results in only a very modest improvement in overall survival.323 Furthermore, a recent analysis of internal documents that became available after the U.S. Food and Drug Administration approval questions the adequacy of the trial and correctness of the conclusions drawn.382
Epithelial Cell Adhesion Molecule. Normal and malignant cells of epithelial origin express the transmembrane glycoprotein epithelial cell adhesion molecule (Epcam).383 Anti-Epcam antibody 17-1A384 showed promising results in patients with colon cancer in several early studies,385 whereas subsequent randomized clinical trials consistently failed to show any benefit.386 A high-affinity engineered anti-Epcam antibody caused cases of acute pancreatitis and was discontinued.387 Other anti-Epcam antibodies in the form of bi- and trifunctional constructs may be more effective, but finding efficacy while keeping lethal toxicity under control presents a major hurdle.
Tumor Antigens Caused by Altered Glycosylation
Aberrant glycosylation and the overexpression of certain carbohydrate moieties is a consistent feature of cancers,388,389 and tumor-associated oligosaccharides are actively investigated as targets for immunotherapy.
Mucins. Mucins are normally heavily glycosylated glycoproteins (ie, containing complex O-glycans) that protect the luminal mucous epithelial surfaces. Mucins of cancer cells often show decreased expression of the complex O-glycans and increased expression of short oligosaccharides, the TF, sialyl-Tn, and Tn antigens390 (see following discussion). Thus, human adenocarcinomas of the pancreas, breast, and colon express mucins391 that can be recognized on cancer cells by MHC-unrestricted cytolytic T cells. These T cells apparently react specifically with repeated epitopes on the protein core of the mucin molecules,392 exposed because of deficient glycosylation in the malignant cells.393 The epitopes are expressed at high density, do not require processing, have a stable conformation, and can directly bind to certain TCRs without being presented by MHC molecules.394 Hypoglycosylated MUC1 is expressed on about two-thirds of newly diagnosed cancers. However, multiple types of vaccines using the nonglycosylated tandem repeat peptides or tumor-associated saccharide antigens conjugated to carriers failed to immunize effectively.395,396 This finding is consistent with multiple lines of evidence that humans and MUC1-transgenic mice are tolerant to the unglycosylated long MUC1 peptide. Numerous approaches have been developed capable of overcoming this tolerance such as using short and long synthetic glycopeptides or plant-expressed MUC1.397,398,399,400 Many different mucin genes have been identified401 and antigens have been defined, but evidence for clinical efficacy with controllable toxicity is lacking.
T and Tn Antigens. Tn antigen on human erythocytes, first described by Moreau et al. in 1957,402 is the cause of a hemolytic autoimmune disorder, Tn syndrome.403 Tn (“T antigen nouvelle”) is a Ser/Thr-O-linked N -acetylgalactosamine monosaccharide distinct from the disaccharide T (or TF) antigen galactose-β1-3-N-acetylgalactose O-linked to a Ser or Thr (Galβ1-3GalNAcα1-Ser/Thr) described earlier by Huebner, Thomsen, and Friedenreich.404 Human cancers very frequently express TF and Tn antigens.405,406 Recently it was shown that the Tn syndrome is caused by somatic mutations in the chaperone COSMC.407 While TF antigen is an oncofetal antigen highly expressed in the embryo and fetus,390 there is no evidence that Tn is also an oncofetal antigen.404 Mutational deletion of the chaperone COSMC leads to expression of the Tn antigen on virtually every embryonic cell and leads to early embryonic death.408 Most adults naturally have anti-Tn as well as anti-TF antibodies due to antigenic stimulation by Tn and TF antigens expressed on the bacterial flora.409,410 Tn antigen is also expressed on human immunodeficiency virus-1 and pathogenic parasites. Thus, evidence is lacking for any expression of Tn antigen on adult or embryonic human or murine cells except by patients suffering from Tn syndrome and cancers that may or may not show a tumor-specific deletion of COSMC. There is no evidence that Tn is an effective therapeutic target. This may be due to anti-Tn antibodies being usually IgM or IgA of very low affinity, though recently developed IgG antibodies reduced slightly the growth rate of human cancer cells in vitro and in vivo.411
Glycopeptide Antigens Resulting from Tumor-Specific COSMC Mutations. The remarkable characteristic of this new class of antigens is the exquisite tumor specificity of the antigen412,413,414 even though it is encoded by normal genes. Appearance of the antigen, however, depends on a tumor-specific somatic mutation that destroys the chaperone COSMC that is essential for any functioning of the core 1β (1-3) galactosyl-transferase, T-synthase. COSMC protects the newly synthesized T synthase from aggregation and subsequent endoplasmic reticulum-associated degradation.404 T-synthase is essential for extending O-linked glycosylation beyond a single O-linked N-acetylgalactosamine (ie, Tn antigen). Importantly, the antigen recognized by the high-affinity, tumor-specific antibody does not bind Tn alone but contains the Tn hapten (ie, the single O-linked N– acetylgalactosamine on a threonine or serine). X-ray crystallography shows that the antibody completely envelops the carbohydrate moiety while interacting with the unique sequence of the peptide moiety in a shallow groove.413 Because Tn does not exist on normal embryonic or adult human or murine cells, there is no central or peripheral tolerance to these antigens allowing high-affinity destructive immune reactions to occur. This also explains the severity of the autoimmune disease associated with the Tn syndrome caused by somatic (not germline) mutations of the COSMC gene.407 Importantly, somatic tumor-specific mutations disabling the X chromosome encoded COSMC gene (for which one copy is naturally silenced) seem to be more common than originally assumed and occur in a wide variety of spontaneous or virally induced human and murine cancers.415 Nevertheless, it must be shown that high-affinity receptors when used as CARs on T cells or linked to anti-CD3 are effective against tumors but have little if any toxicity to
normal tissues. This may be expected from the fact that the epitopes depend on somatic tumor-specific mutations.
normal tissues. This may be expected from the fact that the epitopes depend on somatic tumor-specific mutations.
Oncofetal, Carcinoembryonic, and Cancer-Germline Antigens
Over 50 years ago, human cancer cells were found to express antigens that serologically cross-react with normal embryonic tissue.416 Since then, it has been postulated repeatedly that certain normal genes that are completely silent in all nonmalignant cells may be activated exclusively in malignant cells.417 Alternatively, it has been postulated that cancer cells may express proteins (or immature forms of a protein) that are only expressed in fetal but not in nonmalignant adult cells.418 There certainly is profound hope for finding a universal tumor rejection antigen that can be used for vaccination, prevention of cancer, and antitumor therapy. However, repeated claims of selective activation of normal genes or selective expression of immature forms of these proteins in cancer cells leading to tumor-specific antigens have not been substantiated. Expression of the same antigen by at least one normal cell type in the adult was later often discovered.419,420,421,422
Cancer-Germline/Cancer-Testis Antigens. Changes in gene methylation lead to the expression of a large number of genes encoding a group of antigens referred to “cancer-testis,” “cancer-germline,” or “cancer-spermatogonal” antigens. Many cancer-testis antigens have been described in humans such as MAGE, BAGE, GAGE, LAGE/NY-ESO-1, SAGE, HAGE, and BORIS.422,423,424,425,426,427,428,429,430 All cancer-testis antigens are expressed at high levels in spermatocytes in the testis. And most, if not all, are also expressed in the thymus.430,431,432 Many, but not all, cancer-testis antigens are encoded on the X chromosome.433 Most of cancer-testis antigens can be recognized by autologous cytotoxic T-lymphocyte (CTLs), but there is no evidence for these antigens being generally effective targets for cancer cell destruction in human.434,435 P1A was the first cancer-testis antigen to be identified in the mouse mast cell tumor line P815.436,437 P1A can be induced in a broad range of tumors of diverse histologic origins with the demethylating agent 5-aza-2′-deoxycytidine.438 Various normal tissues including thymus and premeiotic spermatocytes also express P1A.437,439 While active immunization has only modest protective effects against cancer cell inoculation,440 adoptively transferred T cells (monoclonal or polyclonal) targeting only the P1A antigen shrink large tumors (>1 cm in diameter) for several weeks followed by relapse caused by epitope-loss variants.441 For eradication, cancer cells must first be transfected to express costimulatory molecules, which probably leads to the induction of T cells to recognize other tumor antigens.442 Remarkably, autoimmunity has not been reported to be associated with a response to this antigen. In humans, the HLA-A2-restricted NY-ESO1 peptide has very poor affinity to its presenting MHC class I molecule, but high-affinity TCRs to this antigen have been generated and their efficacy in therapy is being explored.443 While many cancer-testis antigens have been described and studied extensively for two decades, their usefulness as targets eradicating human cancers still remains to be shown. Despite these uncertainties, occasional successes have been observed. The hypothetical explanation: the relatively few T cells induced against the cancer-testis antigen served to elicit additional antitumor T-cell clones directed against other antigens that are actually responsible for the tumor regression by a poorly understood phenomenon referred to as epitope spreading.434,444
Numerous cancer-testis antigens have been detected using sera of cancer patients for identifying antigens by recombinant expression cloning (SEREX). Importantly, sera from mice infected with cytopathic or noncytopathic viruses or injected with tumor cell lysates also show an autoantibody response of broad specificity, and intriguingly the majority of the identified autoantigens have been previously described as autoantigens in humans.445,446 This suggests that human SEREX antigens may have to be regarded as afterglows of infection-associated immunopathology and/or tissue damage. This is consistent with the assumption that the human adult IgG autoantibody repertoire is the result of lifelong encounters with bacterial and viral agents and tissue damage. Together, usefulness of the SEREX-defined selfantigens remains to be demonstrated.
Carcinoembryonic Antigen. CEA is a 200-kDa membraneassociated glycoprotein that is expressed not only in fetal but also in adult nonmalignant tissues such as normal colonic mucosa, lung, and lactating breast tissue.447,448 It is released into surrounding fluids. At one time, it was hoped that CEA could be used as a marker for early diagnosis of gastrointestinal and other malignancies449; however, elevated serum levels of CEA are also found in the absence of malignancy (eg, in smokers and in inflammatory bowel diseases such as ulcerative colitis). Though serum levels of CEA are not useful for detecting early cancer, the level of CEA in the blood can be used to monitor the effects of therapy to indicate whether a cancer has been successfully eradicated or has recurred.450 Using CEA as immunotherapeutic target is highly problematic because of severe toxicity. Mice expressing the target antigen in normal tissues closely resembling expression in humans showed severe toxicity from anti-CEA T-cell responses,205 a result that predicted the severe adverse effects later observed in patients treated with anti-CEA CARs.206
Alpha-Fetoprotein. Alpha-fetoprotein (AFP) was the first defined oncofetal protein.451 Though produced by fetal liver and yolk sac cells, it is also present in small amounts in cells and the serum of normal adults. The amount of this protein is elevated in some patients with cancer of the liver or testis and also in some patients with various nonmalignant liver diseases. Therefore, similar to CEA, using AFP as a marker for the early diagnosis of cancer is of questionable use.452 Nevertheless, assays of AFP can detect primary liver cancer at a time when the cancer is treatable, and AFP assays are also used for monitoring patients after therapy.452 The use of AFP as target for active or passive immunotherapy is complicated by lack of specificity.417,453
Clonal Antigens
Clonal antigens are expressed only on the clone of cells from which the cancer originated.454 Except for idiotypes of surface Ig-positive B-cell and T-cell malignancies, there
are presently no candidates for other clonal antigens. Immunization of animals against the idiotype induces an idiotype-specific transplantation immunity against the growth of cancers (myeloma, lymphoma, and leukemia) expressing the idiotype.455,456 Furthermore, idiotype-specific antibodies prevented the growth of surface idiotype-positive murine malignant B cells in mice and guinea pigs in vivo and in vitro,457,458,459,460 and occasionally induced the cancers to go into a long-lasting dormant state.461 Finally, idiotypespecific monoclonal antibodies have caused several partial remissions and one complete remission in patients with B-cell lymphoma,462 and targeting the idiotype on B-cell malignancies continues to be explored.463,464,465 It is necessary to generate different monoclonal antibodies or idiotypic vaccines for each individual cancer to be treated, but the advantage of using such clonal antigens over less restricted tumor-associated antigens is that eliminating the few normal cells bearing the same antigen would probably not adversely affect the patient.
are presently no candidates for other clonal antigens. Immunization of animals against the idiotype induces an idiotype-specific transplantation immunity against the growth of cancers (myeloma, lymphoma, and leukemia) expressing the idiotype.455,456 Furthermore, idiotype-specific antibodies prevented the growth of surface idiotype-positive murine malignant B cells in mice and guinea pigs in vivo and in vitro,457,458,459,460 and occasionally induced the cancers to go into a long-lasting dormant state.461 Finally, idiotypespecific monoclonal antibodies have caused several partial remissions and one complete remission in patients with B-cell lymphoma,462 and targeting the idiotype on B-cell malignancies continues to be explored.463,464,465 It is necessary to generate different monoclonal antibodies or idiotypic vaccines for each individual cancer to be treated, but the advantage of using such clonal antigens over less restricted tumor-associated antigens is that eliminating the few normal cells bearing the same antigen would probably not adversely affect the patient.
Viral Antigens (Encoded by Viral Genes)
Cancer-causing tumor viruses such as SV40, polyoma viruses, human papillomavirus (HPV), hepatitis B virus, hepatitis C virus, human T-lymphotropic virus 1, Epstein-Barr virus (EBV), and other herpes viruses and their antigens are discussed in other parts of the chapter. For discussion of a 70-kDa glycoprotein (gp70)419,420,466,467) and other important antigens468,469,470 encoded by ribonucleic acid (RNA) tumor viruses (eg, murine leukemia virus, maize streak virus,471,472,473,474 and mouse mammary tumor virus475), see Coffin476 and Schreiber.477
IMMUNOGENICITY OF AUTOCHTHONOUS CANCERS
There is the widely held misconception that human cancers are usually less immunogenic than are murine tumors.478 However, convincing evidence indicates that many, probably all, human cancers are antigenic. Similarly, even the most antigenic murine UV-induced “regressor” tumors grow progressively and invariably kill the primary host218,479 (Fig. 47.3). Only transplantation of primary murine tumors into young, syngeneic, immunocompetent, tumor-free recipients reveals their antigenicity and immunogenicity. UV-induced regressors are so immunogenic that they are rejected by naïve immunocompetent hosts. Many experimental cancers require preimmunization to be rejected upon transplantation, and the degree of immunogenicity usually refers to the relative strength of antigen-specific protection a tumor can induce against rechallenge with that tumor. Obviously, the resistance of the host to its autochthonous tumor cannot be great at the time it is clinically apparent and progressively growing.228 Only weak reactions can be detected321 because resistance of the autologous host depends on vaccinations
with the autochthonous tumor following complete tumor removal.269 Antigenicity refers to the antigens on a cancer cell. However, a cancer may have strong antigens yet fail to induce a response (ie, lack immunogenicity). A cancer may also be antigenic and immunogenic but resistant to destruction by the immune response it induces. Thus immunogenicity, antigenicity, and immunosensitivity must be distinguished.
with the autochthonous tumor following complete tumor removal.269 Antigenicity refers to the antigens on a cancer cell. However, a cancer may have strong antigens yet fail to induce a response (ie, lack immunogenicity). A cancer may also be antigenic and immunogenic but resistant to destruction by the immune response it induces. Thus immunogenicity, antigenicity, and immunosensitivity must be distinguished.
 FIG. 47.3. Autochthonous Cancers are Indistinguishable in the Primary Host Regardless of Whether They are Highly Antigenic or Not. Only subsequent transplantation into young syngeneic immunocompetent hosts defines these tumors as regressors or progressors, and shows their antigenicity and immunogenicity. |
Progressively growing autochthonous cancers differ greatly in immunogenicity as determined by responses of syngeneic, tumor-free hosts. Autochthonous tumors induced in mice with UV are among the most immunogenic cancers; transplantation resistance to these so-called regressor tumors appears to be absolute rather than relative. Rejection by normal syngeneic mice is observed without prior immunization, even when the largest testable doses of tumor cells or fragments are used.275 Most MCA-induced fibrosarcomas, unless induced in immunodeficient mice (see following discussion),480 display an intermediate degree of immunogenicity in normal mice.268,269,481 This is shown by the fact that induction of immunologic resistance to most chemically induced tumors requires prior immunization because the initial graft of the tumor generally produces progressive lethal growth.268
Importantly, failure of a cancer to induce a tumor-destructive immune response does not mean it lacks either antigenicity or immunogenicity. (See below discussion of sporadic SV40-induced tumors under Immune Surveillance of Cancer228,301.)
Age and Latency
Experimentally, the length of the latency period of a tumor is usually inversely proportional to the dose of carcinogen. In humans, the doses of (ie, the levels of unintentional exposure to) carcinogens and promoters are believed to be usually relatively low. This means usually a long time is needed for the initiated cells to accumulate the multiple genetic events essential for a premalignant or cancerous lesion. Once the neoplastic cells have reached sufficient numbers to stimulate a response, the host may be too old to respond vigorously. The overwhelming majority of human cancers arise in individuals past 60 years of age,254 and it has been shown repeatedly in humans and mice that the immune response to new antigens declines with age258,482,483,484; this should include antigens on the developing tumors. Thus we do not know the extent of which cancers arising in old individuals may have been selected or retained antigenicity and immunogenicity. In stark contrast, most experimental cancers are induced at an age correlating to that of young middle-aged humans. If 2 years of mouse life roughly correlate to 60 years of human life, then most experimental cancers mirror cancers developing in humans 30 years old or less. Tumors induced by MCA or transgenic oncogenes are usually produced in mice less than 1 year old.485,486,487,488,489 The length of the latency period of these MCA-induced tumors correlates inversely with the degree of immunogenicity.229,490,491,492 There is no such correlation in UV-induced tumors275,479 that begin to develop past 9 months of age, mostly in mice more than 1 year old. Old mice beginning at about 9 months of age fail to reject various types of highly antigenic cancer cells that are regularly rejected by young mice.255,256,257,258 Even though UV irradiation is immunosuppressive, young UV-irradiated mice remain immunocompetent long enough to select for antigen loss variants.235 This indicates that advanced age must contribute to allowing the developing cancers to remain immunogenic. An intriguing question is how many of the tumors that spontaneously arise in older animals would grow in younger syngeneic hosts.480 Considerable but only indirect evidence makes it very unlikely that all tumors would grow.
Spontaneous versus Induced
“Spontaneous” cancers that develop without any known exposure to carcinogens tend to be less immunogenic than cancers induced by DNA tumor viruses or by deliberate exposure to carcinogen.266,267,493,494,495 Unfortunately, serially transplanted tumors were used for most of these comparisons, and transplantation may have selected for less immunogenic variants, thereby confusing the results. If spontaneous murine cancers more closely resembled human cancers, this could suggest that human cancers are poorly immunogenic.495 However, most human cancers are not “spontaneous” but induced by environmental carcinogens, have never been transplanted, and develop mostly in old individuals that may not select for cancer variants. Most, if not all, carcinogens are mutagens17 and probably always cause the expression of tumor-specific antigens. However, it is not clear why tumors induced with the same dose of chemical or physical carcinogen may exhibit quite different degrees of immunogenicity.268,479 One reason might be that the actual local dose of carcinogen that is delivered to a particular target cell or target tissue may vary greatly from animal to animal. Another reason might be that mutations are selected that favor malignant behavior irrespective of the degree of immunogenicity of that mutant protein. This is consistent with the observation that considerable differences in immunogenicity of primary UV-induced tumors only become apparent after transplantation into secondary hosts218,479 (see Fig. 47.3).
“Regressors” from Immunocompromised Hosts
Immunocompetence of the host may not affect the cancer incidence but still influence the immunogenicity of the developing tumor. Conversely, immunosuppression or immune deficiency of the host during carcinogenesis should allow growth of highly immunogenic tumors in the absence of such selection.
Clinical Evidence
Clinical support comes from the appearance of highly antigenic EBV-associated lymphomas in immunosuppressed transplant recipients that are virtually unknown to occur in immunocompetent humans.502 In addition, renal transplant patients experience a reduced risk of developing UV-induced skin cancer after immunosuppressive medications are stopped.503 It is tempting to suggest that many of these
cancers developing in transplant patients are actually “regressor” tumors that have arisen only because host defenses were damaged.
cancers developing in transplant patients are actually “regressor” tumors that have arisen only because host defenses were damaged.
Experimental Evidence
In normal adult mice, Moloney sarcoma virus496 induces tumors larger than 2 cm in diameter that then regress while tumors continue to grow and kill immunodeficient adult or newborn mice.497,498,499 The first experimental evidence for UV-induced regressors came from Kripke in 1974,275 showing that UV irradiation made mice immunodeficient and induced cancers that were often “regressors,” meaning that they were rejected with any testable size inoculum by naïve syngeneic immunocompetent mice. Tumor transplants will grow for about a week and then disappear, though small numbers of the same tumor cells will grow and kill athymic nude mice. The same regressors grew after inoculation of few cancer cells in UV-irradiated mice. So, it may be highly relevant that some carcinogens are immunosuppressive.500,501 Repeated exposures of mice to UV induce persistent immune suppression,502,503,504 leading to the development of highly immunogenic regressor tumors. In contrast, the single injection of MCA induces tumors in 100% of mice and only a short-lived state of immune suppression,501 thus allowing a fully immunocompetent host to select for less immunogenic variants. The concept that immunocompetence of a host influences the immunogenicity of the developing tumor has been tested experimentally by Roberts and Daynes480 comparing the MCA-induced tumors occurring in immunodeficient UV-irradiated mice with cancers induced in normal mice. Indeed, cancers induced with MCA in mice immunosuppressed with UV were frequently regressor tumors, whereas none of tumors induced with MCA in immunocompetent mice were regressors.480 Decades later, this concept was confirmed with tumors induced with MCA in nude, severe combined immunodeficiency, or Rag-/- mice.487,505,506
Selection by the Immunocompetent Host
Immunocompetent hosts can select for cancer variants. Thus, when regressors are transplanted into normal immunocompetent hosts, heritable progressor variants can escape. Some of these variants show antigen loss,218,219,507,508,509 but many others retain their antigenicity and grow faster than the parental tumor when their growth is compared in T, B, or NK cell-deficient hosts,43,45,218,307,505,508,510 suggesting that mechanisms other than antigenicity must participate determining the differences in growth behavior between regressors and progressors.
“Regressors” from Immunocompetent Hosts
Human cancers are sporadic (ie, occur at irregular times and locations); this is in part due to the occasional genetic events that can cause malignant transformation in a single cell that expands to become a detectable tumor. A murine model of sporadic genetic events is the recombinational activation of a normally silenced transgene encoding the strongly oncogenic and strongly antigenic molecule SV40 T, thereby causing sporadic appearance of cancers.228,301 These tumors are regressors. The tumors “snuck” through immune surveillance and immunoselection despite expressing such a powerful antigen and oncogene, but not unnoticed. The developing cancers did induce antibodies and proliferation of antigen-specific T cells during their early dormant phase of growth, but developing tumor-specific T cells were anergized in that they failed to produce interferon (IFN)γ and kill tumor cells in vivo. In contrast, titers of the T antigen-specific IgG response increased with progressive tumor growth.
EFFECTOR MECHANISMS IN CANCER IMMUNITY
Because cancer is not a single disease, it is not surprising that findings using one tumor model may not apply to other tumor models. Considering the antigenic diversity found in tumors, it is also not surprising that different types of innate and adaptive, humoral and cell-mediated immunity have been shown to play different roles in the destruction or enhancement of malignant cells in one or another of the numerous tumor models.
Assays to Study Effector Mechanisms in Vivo
In principle, five different assays have been used to evaluate the importance of different effector mechanisms in vivo. The first type of assay involves transfer of effector cells, cytokines, or antibodies into sublethally irradiated, cyclophosphamide-pretreated, or normal animals challenged with tumor cells. There are certain limitations of this assay. Effector cells or molecules may not reach or localize in the tumor unless both the effector cells and cancer cells are injected intravenously, and both may be trapped in the lungs. Furthermore, if transferred cells or reagents are effective, the assay does not rule out that other effector mechanisms of the host may have been activated by the procedure. In a second procedure, called the Winn assay,229,269,511 tumor cells are mixed with effector cells or serum in vitro; the mixture is injected subcutaneously into an animal to determine whether tumor growth in vivo is prevented. Tumor cells may be killed within minutes before or shortly after the injection, though the readout takes much longer. Therefore, the Winn assay is, in part, an in vitro cytotoxicity assay, even though the host is used as a readout for viable cancer cells. A third method involves elimination of specific lymphocyte subsets or cytokines in vivo by treatment with antibodies specific for different lymphocyte subsets or cytokines. Failure of the host to resist a tumor challenge indicates that the particular subsets or cytokines are an essential component of the host resistance. A fourth method is to use mice genetically deficient in a certain effector mechanism, cell, or cytokine. An analysis of tumor variants that have escaped tumor destruction by the host provides a fifth way for determining the importance of immunologic effectors in vivo.43,44,45,218,235,236,507,512 The phenotypic changes observed in these variants may indicate which effector mechanism was responsible for the selection. Therefore, the type of phenotypic change
may give insight into the relative importance of a naturally occurring defense mechanism that may function in immunocompetent mice (analogous to deducing the action of an antibiotic from the type of change found in the bacterium that has become drug resistant). Host-selected variants from experimentally induced regressor tumors are used extensively for this approach.
may give insight into the relative importance of a naturally occurring defense mechanism that may function in immunocompetent mice (analogous to deducing the action of an antibiotic from the type of change found in the bacterium that has become drug resistant). Host-selected variants from experimentally induced regressor tumors are used extensively for this approach.
Antibodies and B Cells
The role of B cells in regulating tumor immunity continues to be studied extensively yet remains poorly understood. In a tumor model of leukemia in which CD4+ helper T cells were required for successful treatment, B cells were necessary for efficient T-cell priming.513 Conversely, in other tumor models, elimination of CD4+T cells promoted tumor rejection by CD8+ cells, and an absence of B cells improved CTL responses and tumor rejection.514,515,516 Antibodies and B cells have been associated with enhanced papilloma formation and autochthonous cancer development.341,489,517,518,519 The mechanism recently postulated is that regulatory B cells help the macrophage become tumor-promoting,520,521 or that they suppress surveillance by T cells that normally eliminate neoplastic cells with oncogenic mutations.519 There are other possible mechanisms.342,343 Also, none of these experiments identified the antigens involved, except one study showing that induction of mutant-ras specific-antibodies correlates with much more effective papilloma development in an autochthonous tumor model.341
Human antisera and monoclonal antibodies reactive with autologous tumors have been isolated.420,522 However, a strong humoral response to tumor antigens does not correlate with demonstrable resistance of the host to the tumors. TL+ leukemias induce high titers of TL-specific antibodies that are cytotoxic to TL+ leukemia cells in vitro in the presence of heterologous complement,420 but TL+ leukemias grow equally well in immunized mice having high titers of TL antigen-specific antibody and in nonimmunized mice.523,524 Similarly, humoral immune response to MCA-induced sarcomas does not provide protective immunity against a tumor transplant.525 Obviously, the presence of antibodies for these kinds of tumors has no relevance in predicting whether the host will reject the tumor.
Related posts:
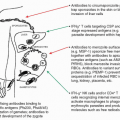
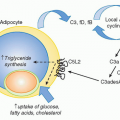
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


