Chapter Outline
CLINICAL APPLICATION OF CHEMOTHERAPY
PHARMACOKINETIC CHANGES DURING CHILDHOOD
PHARMACOGENETICS AND PHARMACOGENOMICS OF CHEMOTHERAPEUTIC AGENTS IN CHILDREN
STRATEGIES TO IMPROVE THE THERAPEUTIC INDEX
Dosage Based on Body Surface Area
Dose Adaptation Using Pharmacokinetic-Pharmacodynamic-Pharmacogenetic Principles
In the past two decades we have seen substantial advances in the number of available chemotherapeutic agents to treat a variety of neoplastic diseases, as well as in the discovery and development of new anticancer drugs, while our understanding of the biology and genetics of cancer continues to grow. The tools at our disposal now include classes of molecules with highly distinct structures and innovative mechanisms of action and an impressive array of new techniques and novel uses for existing agents. In this chapter we will review the principles of chemotherapy in children, discuss the factors that influence individual responses to chemotherapy, and describe how pharmacokinetic, pharmacodynamic, and/or pharmacogenetic principles are used when conducting clinical trials that provide therapy tailored to individual patients.
Chemotherapeutic Agents
Chemotherapeutic agents used to treat pediatric cancer are generally similar to those used with adults, including conventional cytotoxic drugs and molecularly targeted agents. The former class can be subdivided into two large groups based on the dependence of their mechanism of action on the cell cycle. Cell cycle–nonspecific agents, which include alkylating agents, platinum agents, and most antitumor antibiotics, kill both quiescent and cycling tumor cells. Cell cycle–specific agents include antimetabolites and agents that interact with tubulin and topoisomerases. Conventional cytotoxic agents can target both malignant and normal cells and often cause adverse effects. During the past decade, the development of molecularly targeted chemotherapy has been revolutionized by a greater understanding of the biologic and genetic features of malignant disease. Table 46-1 provides an overview of the drugs used to treat childhood cancers and their key pharmacologic features.
| Drug (Trade Name; Chemical Class) | Mechanism of Action | Antitumor Activity | Route of Administration | Primary Pathway of Elimination | Enzymes and Transporters | Principal Adverse Effects |
|---|---|---|---|---|---|---|
| Alkylating Agents | ||||||
| Mechlorethamine (Mustargen; nitrogen mustard, mustine, HN2) | Alkylation; DNA cross-linking | Hodgkin lymphoma (MOPP) | IV injection | Hydrolysis; liver | N -demethylation (CYP) | A, M, N&V, Mu, HU, Derm, V, Phleb, G, SM |
| Melphalan (Alkeran; mechlorethamine analogue) | Alkylation; DNA cross-linking | HDCT | IV injection or infusion; oral | Hydrolysis; renal | GSTP1, LAT1, ABCB1 | A, M, N&V, D, Mu, Derm, Vas, Phleb, SIADH, G, SM |
| Chlorambucil (Leukeran; mechlorethamine analogue) | Alkylation; DNA cross-linking | Hodgkin lymphoma (ChlVPP) | Oral | Hydrolysis | GSTP1, ABCC2 | A, M, N&V, HU, P, S, G, SM |
| Cyclophosphamide (Cytoxan; mechlorethamine analogue) | Alkylation (prodrug); DNA cross-linking | Leukemias, lymphomas, neuroblastoma, sarcoma | IV infusion; oral | Hydrolysis; liver | CYP2B6, CYP2C9, CYP3A4, CYP3A5, GSTA1, GSTP1, ALDH1A1, ALDH3A1 | A, M, N&V, Cy, SIADH, C, P, G, SM |
| Ifosfamide (Ifex; mechlorethamine analogue) | Alkylation (prodrug); DNA cross-linking | Sarcomas, germ cell | IV infusion | Hydrolysis; liver | CYP2A6, CYP2B6, CYP3A4, CYP2C19, OCT2 | A, M, N&V, Met, Cy, SIADH, NT, C, P, R, G, SM |
| Bendamustine (Treanda) | Alkylation; DNA cross-linking; blocks purine synthesis | Hodgkin lymphoma, lymphoma, leukemia | IV infusion | Liver | CYP1A2 | M, N&V, H, P, PE, NT |
| Thiotepa (Thioplex; aziridine) | Alkylation; DNA cross-linking | HDCT | IV injection or infusion; intrathecal, intravesical | Liver | NA | A, M, N&V, HU, Derm, Cy, G, SM |
| Busulfan (Myleran; alkyl alkane sulfonate) | Alkylation; DNA cross-linking | HDCT | IV infusion; oral | Conjugation with glutathione; liver | CYP3A4, GSTA1, GSTP1 | A, M, N&V, Mu, Derm, HU, AI, NT, VOD, P, S, G, SM |
| Carmustine (BiCNU; BCNU, nitrosourea) | Alkylation; DNA cross-linking; carbamoylation | Brain tumors, Hodgkin lymphoma, lymphoma | IV infusion; wafer implant (glioblastoma) | Chemical hydrolysis; liver | CYP | M, N&V, Phleb, S (wafer), C, H, P, R, G, SM |
| Lomustine (CeeNU; CCNU, nitrosourea) | Alkylation; DNA cross-linking; carbamoylation | Brain tumors, Hodgkin lymphoma, lymphoma | Oral | Chemical hydrolysis; liver | CYP2D6 | M, N&V, NT, C, H, P, R, G, SM |
| Dacarbazine (DTIC, DTIC-Dome; imidazole-carboxamide) | Methylation inhibition (prodrug) | Hodgkin lymphoma, neuroblastoma, sarcomas | IV infusion | Chemical hydrolysis; liver, renal | N-demethylation (CYP); CYP1A2, CYP2E1 | M, N&V, Flu, H |
| Procarbazine (Mutulane; imidazole-carboxamide) | Methylation inhibition (prodrug) | Brain tumors; Hodgkin lymphoma (ChlVPP) | Oral | Chemical hydrolysis; liver, renal | CYP, MAO | M, N&V, Mu, Flu, NT, Derm, G, SM |
| Temozolomide (Temodar; imidazole-carboxamide) | Methylation inhibition (prodrug) | Brain tumors | Oral | Chemical hydrolysis | CYP (minor) | M, N&V, NT, PE, H |
| Antimetabolites | ||||||
| Cytarabine (Cytosar; Ara-C, cytosine arabinoside; cytidine analogue) | Incorporated into DNA; inhibits DNA polymerase; terminates DNA chain elongation | Leukemias | IV injection or infusion; intrathecal; SC | Activated to triphosphate; deamination | Cytidine deaminase, CNT1, ENT1, ENT2, ABCC4, ABCC11 | M, N&V, Mu, GI, HU, Derm, HFS, PE, Conj, NT, H, PE |
| Azacytidine (Vidaza; cytidine analogue) | Hypomethylation of DNA (low dose) | MDS, AML | SC | Activated to triphosphate; chemical degradation; deamination | Cytidine deaminase | M, N&V, F, Mu, D, Inj, Derm |
| Decitabine (Dacogen; cytidine analogue) | Hypomethylation of DNA (low dose) | MDS, AML | IV infusion | Activated to triphosphate; chemical degradation; deamination | Cytidine deaminase | M, N&V, F, Mu, D, Derm |
| Gemcitabine (Gemzar; cytidine analogue) | Incorporated into DNA; inhibits DNA polymerase; terminates DNA chain elongation; inhibits ribonucleotide reductase (prodrug) | Solid tumors, lymphoma | IV infusion | Activated to triphosphate; deamination | Cytidine deaminase, CNT1, ENT1 | M, N&V, Mu, D, F, Flu, Derm, NT, PE, H, R |
| Cladribine (Leustatin; adenosine analogue) | Incorporated into DNA; inhibits DNA polymerase; terminates DNA chain elongation; inhibits ribonucleotide reductase (prodrug) | Leukemias | IV infusion | Activated to triphosphate; renal; liver | Oxidative cleavage, oxidation, conjugation, CNT1, CNT2, CNT3, ENT1, ABCG2 | M, N&V, D, Derm, NT |
| Clofarabine (Clolar; adenosine analogue) | Incorporated into DNA; inhibits DNA polymerase; terminates DNA chain elongation; inhibits ribonucleotide reductase (prodrug) | Leukemias | IV infusion | Activated to triphosphate; extrahepatic | NA | M, N&V, D, C, H, R |
| Fludarabine (Fludara; adenosine analogue) | Incorporated into DNA; inhibits DNA polymerase; terminates DNA chain elongation; inhibits ribonucleotide reductase (prodrug) | AML, CML, indolent lymphomas | IV infusion | Activated to triphosphate; renal | NA | M, HA, Derm, fever, H, NT, TLS |
| Nelarabine (Arranon) | Incorporated into DNA; inhibits DNA synthesis | ALL | IV infusion | Activated to ara-guanine triphosphate; liver, renal | Adenosine deaminase | M, N&V, NT, P, PE |
| 6-Mercaptopurine (Purinethol; hypoxanthine analogue) | Incorporated into DNA, RNA; blocks purine synthesis (prodrug) | ALL | Oral | Liver, renal (high dose) | TPMT, HGPRT, ENT2, OAT2, OAT3, ABCC4 | M, H, Mu, N&V, HU, crystalluria, GI |
| 6-Thioguanine (guanine analogue) | Incorporated into DNA, RNA; blocks purine synthesis (prodrug) | ALL, AML | Oral | Liver | TPMT, HGPRT, CNT3, ENT2, OAT3, ABCC5 | M, H, Mu, N&V, HU, crystalluria |
| Hydroxyurea (Hydrea, Droxia) | Interferes with synthesis of DNA synthesis; inhibits ribonucleoside diphosphate reductase | ALL, AML | Oral | Liver, renal | OATP1B3, UT-A, UT-B1 | M, H, Mu, N&V |
| 5-Fluorouracil (fluoropyrimidine) | Incorporated into RNA, DNA; inhibits thymidine synthase (prodrug of 5′-DFUR) | Carcinomas, hepatic tumors | IV injection or infusion | Liver | DPD, GSTP1, hNT1, OAT2, | M, Mu, N&V, HFS, D, Derm, NT, C |
| Capecitabine (Xeloda; fluoropyrimidine) | Incorporated into RNA, DNA; inhibits thymidine synthase (prodrug of 5′-DFUR) | Colorectal cancer (in development) | Oral | Urine | CES2, CES1A1, cytidine deaminase, thymidine phosphorylase, DPD, CNT1, ABCA1 | M, Mu, N&V, HFS, D, Derm, NT, C |
| Methotrexate (Trexall; antifolate) | Interferes with folate metabolism | Leukemias, lymphomas, osteosarcoma | Oral; IM; SC; IV injection; intrathecal | Renal | FPGS, GGH, OAT1, OAT2, OAT3, OAT4, OATP1A2, OATP1B1, OATP1B3, OATP4C1, RFC, PCFT, ABCB1, ABCC1, ABCC2, ABCC3, ABCC4, ABCG2 | M, Mu, Derm, H, R, NT |
| Pemetrexed (Alimta; antifolate) | Interferes with folate metabolism | Various (in development) | IV infusion | Renal | NA | M, fatigue, Mu, HFS, Derm |
| Antitumor Antibiotics | ||||||
| Bleomycin (Blenoxane) | Free radical–mediated DNA strand breaks | Lymphomas, germ cell tumor | IV injection; IM, SC | Renal | OCT6 | P, Derm, fever, Mu, A, HSR, N&V |
| Dactinomycin (Cosmegen) | DNA cross-linking | Solid tumors | IV injection | Renal, liver | NA | M, Mu, N&V, D, A, H, V |
| Mitomycin C (Neulasta) | DNA cross-linking | Carcinomas | IV injection or infusion; intravesical | Liver | ABCB1 | M, N&V, Mu, HU, anemia, VOD |
| Daunorubicin (Daunomycin; anthracycline) | DNA strand breaks (topo II) | ALL, AML, lymphomas | IV injection or infusion | Renal, liver | OCTN1, ABCB1, aldoketoreductase | M, Mu, N&V, D, A, C, V |
| Doxorubicin (Adriamycin; anthracycline) | DNA strand breaks (topo II) | ALL, AML, lymphomas, solid tumors | IV infusion | Liver | CYP2D6, CYP3A4, GSTP1, OCT6, OCTN1, ABCB1, ABCB5, ABCG2, aldoketoreductase | M, Mu, N&V, A, D, C, V |
| Epirubicin (Ellence; anthracycline) | DNA strand breaks (topo II) | Solid tumors, leukemia (in development) | IV infusion | Renal, liver | ABCB1, ABCC1, ABCG2, aldoketoreductase | M, Mu, N&V, A, C, V |
| Idarubicin (Idamycin; anthracycline) | DNA strand breaks (topo II) | ALL, AML, lymphomas | IV injection or infusion; oral | Renal, liver | ABCB1, aldoketoreductase | M, Mu, N&V, D, A, C, V |
| Mitoxantrone (Novantrone; anthracenedione) | DNA strand breaks (topo II) | ALL, AML, lymphomas | IV infusion; IP | Renal, liver | OCTN1, ABCB1, ABCG2 | M, Mu, N&V, A, urine, veins and nail discoloration |
| Asparaginases | ||||||
| l-Asparaginase (Elspar, Kidrolase; native asparaginase from Escherichia coli ) | Depletion of plasma asparagine | ALL, lymphomas | IV infusion (>30 min), IM | Nonrenal | NA | HSR, thrombosis, GI, glucose intolerance, coagulopathy, hyperglycemia, H |
| Crisantaspase (Erwinase; Erwinia l-asparaginase, native asparaginase from Erwinia chrysanthemi ) | Depletion of plasma asparagine | ALL, lymphomas | IV infusion (>30 min), IM | Nonrenal | NA | HSR, thrombosis, GI, glucose intolerance, coagulopathy, hyperglycemia, H |
| Pegasparaginase (Oncaspar; PEG-asparaginase, from E. coli ) | Depletion of plasma asparagine | ALL, lymphomas | IV infusion (1-2 hours) | Nonrenal | NA | HSR, thrombosis, GI, glucose intolerance, coagulopathy, hyperglycemia, H |
| Corticosteroids | ||||||
| Dexamethasone (Decadron) | Receptor-mediated lympholysis | Leukemias, lymphomas | Oral, IV injection; IM | Liver | CYP3A4, ABCB1 | Muscle weakness, osteoporosis, Derm, hypertension, N&V, headache |
| Prednisolone (Orapred OTD, Pediapred) | Receptor-mediated lympholysis | Leukemias, lymphomas | Oral; IV injection | Renal, liver | CYP3A4, sulfation, glucuronidation | Muscle weakness, osteoporosis, Derm, hypertension, N&V, headache |
| Prednisone (Deltasone) | Receptor-mediated lympholysis (prodrug) | Leukemias, lymphomas | Oral | Renal, liver | CYP3A4, sulfation, glucuronidation | Muscle weakness, osteoporosis, Derm, hypertension, N&V, headache |
| Platinum Compounds | ||||||
| Carboplatin (Paraplatin) | Platination; DNA cross-linking | Brain tumors, neuroblastoma, sarcomas | IV infusion | Renal | CTR1, ATP7A | M, N&V, NT, EA, HSR |
| Cisplatin (Platinol) | Platination; DNA cross-linking | Testicular, brain tumors, osteosarcoma, neuroblastoma | IV infusion | Renal | GSTP1, OCT1, OCT2, CTR1, ABCC2, ABCC4, ATP7B, MATE1 | N&V, R, NT, M, EA, O, HSR |
| Oxaliplatin (Eloxatin) | Platination; DNA cross-linking | Colorectal cancer, lymphoma (in development) | IV infusion | Renal | GSTP1, OCT1, OCT2, OCT3, OCTN1, OCTN2, MATE1, MATE2-K | NT, N&V, D, M, R, HSR |
| Retinoids | ||||||
| All- trans -retinoic acid (ATRA; Vesanoid; tretinoin; retinol derivative) | Differentiation agent | Acute promyelocytic leukemia | Oral | Liver | CYP2C8, CYP2A6, CYP2B6, CYP2C9 | Retinoic acid syndrome, pseudotumor cerebri, cheilitis |
| 13- cis -retinoic acid (Accutane; retinol derivative) | Differentiation agent | Neuroblastoma | Oral | Liver | CYP2B6, CYP2C8, CYP2C9, CYP3A4/5, CYP2A6, CYP2D6 | Cheilitis, conjunctivitis, dry mouth, xerosis, pruritus, headache |
| Topoisomerase-Interactive Agents | ||||||
| Etoposide (VePesid; VP-16) | DNA strand breaks (topo II) | ALL, AML, lymphomas, neuroblastoma, sarcomas, brain tumors | IV infusion | Renal, liver | CYP3A4, CYP3A5, CYP1A2, CYP2E1, UGT1A1, OCTN2, OATP1B1, ABCB1, ABCC1, ABCC2 | M, N&V, hypotension, HSR, SM |
| Teniposide (Vumon; VM-26) | DNA strand breaks (topo II) | ALL | IV infusion; oral | Liver | CYP3A4, ABCB1 | M, N&V, HSR, hypotension, SM |
| Irinotecan (Camptosar; CPT-11; camptothecin analogue) | DNA strand breaks (topo I; prodrug of SN-38) | Rhabdomyosarcoma, solid tumors | IV infusion | Renal, liver | CYP3A4, CYP3A5, CYP2B6, CES2, UGT1A1, OATP1B1, ABCB1, ABCC2, ABCC4, ABCG2, | M, D, N&V, A, H |
| Topotecan (Hycamptin; camptothecin analogue) | DNA strand breaks (topo I) | Neuroblastoma; rhabdomyosarcoma | IV infusion | Renal | OAT3, ABCB1, ABCC4, ABCG2 | M, Mu, D, N&V, A, Derm, H |
| Tubulin-Interactive Agents | ||||||
| Docetaxel (Taxotere; taxanes) | Microtubule depolarization inhibitor | Solid tumors | IV infusion | Liver | CYP3A4/5, CYP2C8, OAT2, OATP1B3, ABCB1, ABCC2, ABCC10 | M, HSR, A, NT, Derm, Mu |
| Paclitaxel (Taxol; Abraxane; taxane) | Microtubule depolarization inhibitor | Solid tumors | IV infusion | Liver | CYP2C8, CYP2C9, CYP3A4/5, OAT2, OATP1B1, OATP1B3, ABCB1, ABCC2, ABCC10 | M, HSR, A, NT, M, C |
| Vinblastine (Velban; Vinca alkaloid) | Microtubule polarization inhibitor | Histiocytosis, Hodgkin lymphoma, testicular cancer | IV push or infusion | Liver | CYP3A4, CYP2D6, ABCB1, ABCC1, ABCC2, ABCC10, ABCG2 | M, A, Mu, NT, V |
| Vincristine (Oncovin; Vinca alkaloid) | Microtubule polarization inhibitor | ALL, lymphomas, solid tumors | IV injection | Liver | CYP3A5, CYP3A4, ABCB1, ABCC1, ABCC2, ABCC10 | NT, A, SIADH hypotension, V |
| Vinorelbine (Navelbine; Vinca alkaloid) | Microtubule polarization inhibitor | Solid tumor, Hodgkin lymphoma (in development) | IV injection of infusion | Liver | CYP3A4, CYP2D6 | M, NT, A, V |
| Tyrosine Kinase Inhibitors | ||||||
| Imatinib (Gleevec; STI-571, benzamide analogue) | Abl inhibitor | Ph+ CML, ALL | Oral | Liver | CYP3A4, CYP3A5, CYP2D6, CYP2C9, CYP2C19, OATP1A2, OATP1B3, OCT1, ABCB1, ABCC1, ABCC4, ABCG2 | N&V, F, D, H, M |
| Dasatinib (Sprycel; carboxamide analogue) | Src-Abl inhibitor | Ph+ CML, ALL | Oral | Liver | CYP3A4, FMO3, OATP1B3, ABCB1, ABCC4, ABCG2 | M, D, N&V, Derm, fatigue, P, headache |
| Nilotinib (Tasigna) | Abl inhibitor | Ph+ CML, ALL | Oral | Liver | CYP3A4, OATP1B1, OATP1B3, ABCB1, ABCG2 | M, N&V, D, C, Derm, MS, P |
| Ponatinib (Iclusig) | Src-Abl inhibitor | Ph+ CML, ALL (in development) | Oral | Liver | CYP3A4, CYP2C8, CYP2D6, CYP3A5 | M, N&V, D, C, Derm, H, GI, MS |
| Sunitinib (Sutent; carboxamide analogue) | VEGFR, PDGFR inhibitor | In development | Oral | Liver | CYP3A4, CYP1A2, OATP1B1, ABCB1, ABCG2 | M, D, Derm, anorexia, hypertension, fatigue, headache |
| Sorafenib (Nexavar; carboxamide analogue) | Raf and FLT3 inhibitor | In development | Oral | Liver | CYP3A4, UGT1A9, OATP1B1, OATP1B3, ABCB1, ABCG2 | M, D, Derm, anorexia, hypertension, fatigue, headache |
| Erlotinib (Tarceva; OSI-774, quinazolinamine analogue) | EGFR inhibitor | Solid tumors (in development) | Oral | Liver | CYP3A4/5, CYP1A2, ABCG2 | Derm, D, anorexia, fatigue, N&V, dyspnea, fatigue |
| Gefitinib (Iressa; ZD1839, anilinoquinazoline analogue) | EGFR inhibitor | Solid tumors (in development) | Oral | Liver | CYP3A4/5, CYP2D6, OATP1B3, ABCB1, ABCG2 | D, Derm, N&V |
| Crizotinib (Xalkori) | ALK inhibitor | ALCL, non–small cell lung cancer, neuroblastoma (in development) | Oral | Liver, Renal | CYP3A4/5, OATP1B1, OATP1B3, ABCB1 | Ocular, N&V, Edema, H, NT |
| Pazopanib (Votrient) | Vascular endothelial growth factor inhibitor | Solid tumors (in development) | Oral | Liver | CYP3A4, CYP1A2, CYP2C8, OATP1B1, OATP1B3, ABCB1, ABCG2 | D, N&V, M, H, C, MS, P |
| Vemurafenib (Zelboraf) | BRAF inhibitor | Melanoma | Oral | Liver | CYP3A4, OATP1B3, ABCB1 | Derm, N&V, MS, PE |
| Proteasome Inhibitors | ||||||
| Bortezomib (Velcade) | Proteasome inhibitor | Leukemia, lymphoma (in development) | IV injection, SC | Liver | CYP2C19, CYP3A4, CYP1A2 | M, N&V, NT, D, P |
| Histone Deacetylase Inhibitors | ||||||
| Vorinostat (Zolinza) | Histone deacetylase inhibitor | Leukemia, lymphoma (in development) | Oral | Liver, Renal | NA | D, N&V, PE, A, M, NT, R |
| Other Molecular Targeting Agents | ||||||
| Arsenic trioxide (Trisenox) | Degrades PML-RARA and induces apoptosis | Acute promyelocytic leukemia, MDS | IV infusion | Hydrolysis | NA | C, NT, Derm, N&V, D, M, H, P |
| Everolimus (Afinitor) | mTOR kinase inhibitor | Solid tumor, leukemia, subependymal giant cell astrocytoma (in development) | Oral | Liver | CYP3A4, ABCB1 | N&V, D, HR, P, PE, Derm, M, Mu |
| Temsirolimus (Torisel) | mTOR kinase inhibitor | Solid tumor, leukemia (in development) | IV infusion | Liver | CYP3A4, ABCB1 | N&V, D, HR, P, PE, Derm, M, Mu |
| Ruxolitinib (Jakavi) | Janus kinase inhibitor | In development | Oral | Liver | CYP3A4 | M, D, N&V, H, P, PE |
| Vismodegib (Erivedge) | Hedgehog inhibitor | In development | Oral | Liver | CYP2C9, CYP3A4, ABCB1 | M, A, MS |
| Antibodies | ||||||
| Rituximab (Rituxan) | CD20 antibody | Leukemia, lymphoma | IV infusion | NA | NA | M, N&V, F, PE, NT |
| Gemtuzumab ozogamicin | CD33 antibody, DNA double-strand break | AML | IV infusion | NA | NA | M, H, C, F, N&V, R |
| Brentuximab vedotin (Adcetris) | CD30 antibody, disrupt microtubule network | Hodgkin lymphoma, ALCL | IV infusion | Liver | CYP3A4 | NT, F, PE, Derm, N&V, D, M, P |
| Alemtuzumab (Campath) | CD52 antibody | Leukemia, lymphoma, HSCT | IV infusion, SC | NA | NA | F, M, MS, C |
| Bevacizumab (Avastin) | Vascular endothelial growth factor antibody | Solid tumors (in development) | IV infusion | NA | NA | M, C, D, D, N&V, MS, P, R |
| Biological Response Modulator | ||||||
| Aldesleukin (Proleukin) | Interleukin-2 | Melanoma, leukemia, cell therapy | IV infusion, SC | Renal | NA | D, F, Flu, C, PE, Derm, N&V, M, R, P |
Clinical Application of Chemotherapy
Chemotherapy has four major roles in the management of cancer: (1) primary cytoreduction therapy, (2) adjuvant therapy, (3) neoadjuvant therapy, and (4) site-directed therapy. Primary cytoreduction therapy is used when no superior alternative treatment is available (such as surgery and radiation therapy). Hematologic malignancies and some solid tumors of childhood are significantly chemosensitive and can be cured by serial courses of chemotherapy (e.g., induction, consolidation, and continuation therapy for acute lymphoblastic leukemia [ALL]). The chemosensitive malignancies also include acute myeloid leukemia (AML), lymphomas (both non-Hodgkin and Hodgkin), germ cell tumors, Wilms tumor, and embryonal rhabdomyosarcoma. Chemotherapy can also be used as an adjuvant therapy, wherein systemic treatment is applied to enhance local tumor control and to eradicate occult disseminated disease after surgery and/or radiation therapy. Occult residual disease at original sites and micrometastases are frequently present even after macroscopic resolution of local disease. Before the routine use of adjuvant chemotherapy, 60% to 95% of children with localized solid tumors experienced relapse at primary or metastatic sites after receiving local control therapy alone. Thus the goal of adjuvant chemotherapy is to reduce the risk of local and systemic recurrence by eradicating microscopic residual disease or micrometastases in lungs, bone, bone marrow, lymph nodes, or other sites. The efficacy of adjuvant chemotherapy for Wilms tumor, Ewing sarcoma, rhabdomyosarcoma, osteosarcoma, and brain tumors (e.g., medulloblastoma) is well established. Neoadjuvant therapy may be used to reduce the size of primary and metastatic tumors, allow definitive surgical resection, and eradicate possible micrometastases when localized cancers cannot otherwise be optimally managed. This approach is often used with childhood solid tumors, including osteosarcomas, neuroblastomas, hepatoblastomas, and Wilms tumors. Examples of site-directed therapy include instillation of chemotherapy agents into sanctuary sites, such as intrathecal chemotherapy for leukemia and site-directed perfusion therapy (e.g., injection of agents into the feeding artery of tumors such as sarcomas in the extremities, liver tumors, and retinoblastoma).
Chemotherapy in Children
The current strategy is to administer chemotherapeutic agents at the maximum tolerated dose—that is, the dose just below that which causes unacceptable toxicity. This approach is based on a series of prospective and retrospective analyses showing that the greater the dose intensity (i.e., the dose delivered over a standard time interval), the better the outcome. However, the narrow therapeutic index (i.e., the ratio of the theoretical minimum effective dose to the maximum tolerated dose) of most anticancer drugs demands a rigorous effort to optimize these regimens ( Fig. 46-1 ). Effective systemic treatment requires judicious application of cytotoxic drugs that differ only slightly in their toxicity to normal cells and their therapeutic lethality to malignant cells. Pediatric oncologists and pharmacologists have studied multiple approaches in search of the best way to exploit this narrow therapeutic margin, including the use of drug combinations with different dose-limiting toxicities, the rescue or reconstitution of normal tissues (e.g., administration of leucovorin after methotrexate and use of bone marrow transplantation after high-dose chemotherapy), and the use of biologic modifiers to counteract specific mechanisms of resistance.
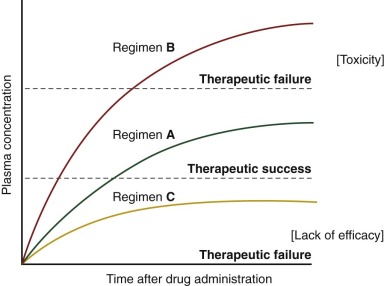
The therapeutic approaches used in adults cannot be directly transferred to growing children because certain agents have an extremely narrow therapeutic range in pediatric patients, and in most cases little or no information is available about the intrinsic sensitivity of a tumor to a particular agent and the tolerability of a given dose. Physiologic differences between children and adults, such as body composition, hepatic metabolism, and renal function, may affect the pharmacokinetics of a drug (i.e., drug disposition involving its absorption, distribution, metabolism, and excretion and the plasma concentration–time profile of the drug in the body) and thus the pharmacodynamics of a drug (i.e., drug action; the quantitative study of the effects of a drug, including efficacy and toxicity). Children may also be more susceptible than adults to the toxicity of agents even when the pharmacokinetics are similar, as in the case of all- trans -retinoic acid for pseudotumor cerebri. Further, pharmacokinetic studies have shown substantial interpatient variability in drug disposition and systemic drug exposure in children; these findings may underlie the observed variability in toxicity and response. The pharmacokinetics and pharmacodynamics of a drug, and thus its anticancer effects, are also influenced by physiologic factors that differ in children and adults, by drug-drug interactions, and by genetic influences (pharmacogenomics). Although pediatric cancer is rare compared with adult cancer, optimizing treatment in a patient group with a higher cure rate and a longer expected survival becomes critical to minimize the incidence of preventable acute and late complications in children while maintaining efficacy.
Pharmacokinetic Changes during Childhood
Pharmacokinetic parameters include half-life (T 1/2 ), area under the plasma concentration versus time curve (AUC), bioavailability, volume of distribution (Vd), steady state concentration, and clearance ( Table 46-2 ). Body composition and organ function at the extremes of age can affect drug disposition and effect. Changes as a child develops and matures may alter absorption, distribution, metabolism, and excretion of chemotherapeutic agents ( Fig. 46-2 ).
| Parameter | Units | Definition |
|---|---|---|
| Half-life (T 1/2 ) | Time (h) | The time necessary to halve the plasma concentration. Useful for determination of frequency of drug administration to obtain the desired plasma concentration. The half-life of a particular drug is usually independent of the dose administered. However, it may change according to the saturation of a mechanism (e.g., elimination, catabolism, or binding to plasma proteins). |
| Area under the curve (AUC) | Concentration × time (µM × h) | Calculated as f ([C] × Dt); [C] is measured concentration and Dt is the interval of time between two measurements. The integral of the plasma concentration versus an interval of definite time. It represents an important quantitative measure of total systemic drug exposure. Also, it is required for the calculation of other pharmacokinetic parameters such as clearance and bioavailability. |
| Bioavailability (F) | Fraction (%) | The percentage of the administered drug concentration that is achieved in the central compartment. Generally measured by comparing the AUCs obtained after intravenous administration with those obtained after oral administration. For example, the AUC obtained after intravenous administration usually corresponds to bioavailability (i.e., 100%). After oral administration, the AUC corresponds at best to an identical bioavailability; it is generally lower. |
| Volume of distribution (Vd) | Volume (L) | The fictitious volume in which the drug would have been distributed by supposing that its concentration is homogeneous (i.e., the average tissue concentration is identical to that of plasma). Expressed as Vd = dose/C0 (initial concentration). For example, after intravenous injection of 100 mg of a drug whose initial concentration, C0, in plasma is 10 mg/L, the volume of distribution is 10 L. For a given drug, the knowledge of its desired concentration in blood and of its volume of distribution allows evaluation of the dose to administer. |
| Steady state concentration (Css) | Concentration (µM) | The state of equilibrium obtained at the end of a certain number of administrations. To obtain an increase in the plasma concentration with repeated administrations, it is necessary that a residual concentration persists at the time of the following administration. At the steady state, if the dose and the frequency of administration remain constant, the concentration obtained will also be constant. The steady state is usually obtained at the end of approximately five half-lives. |
| Clearance (Cl) | Volume/Time (mL/min) | Defined as the theoretic volume from which drug is completely removed per unit time. Plasma clearance is the apparent volume of plasma from which drug is cleared per unit of time. Total clearance is the fraction of the volume of distribution, Vd, which is completely cleared per unit of time. The total clearance depends on the constant of elimination and thus on T 1/2 and on Vd, which is considered to be sum of metabolic and spontaneous chemical degradation, as well as biliary/fecal and renal elimination. |
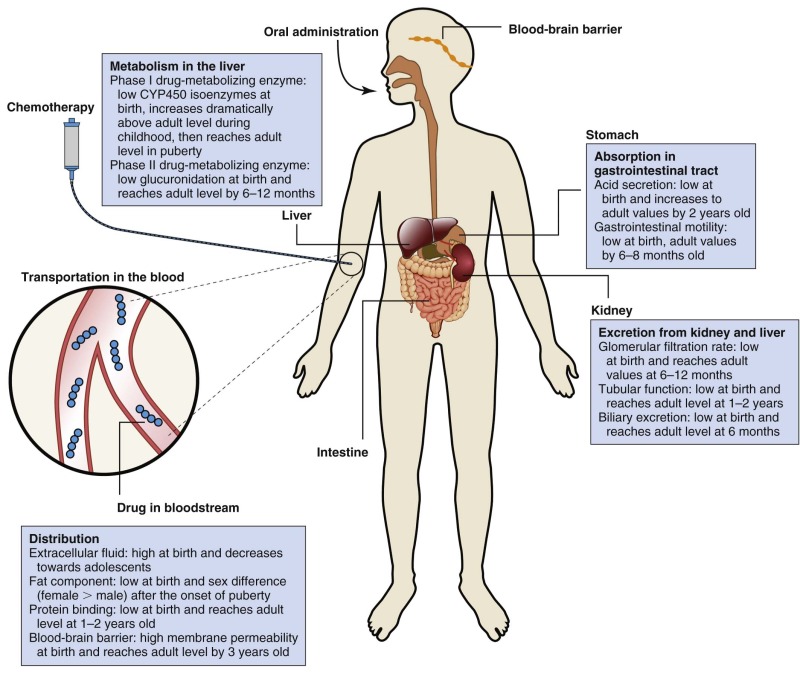
The importance of understanding the influence of age on the pharmacokinetics and pharmacodynamics of individual anticancer agents has increased steadily with advances in the treatment of infant and childhood malignancies. Although the influence of age has been formally evaluated for a limited number of cytotoxic drugs, further efforts to improve the individualization of drug therapy on the basis of maturation and development are necessary to enhance the odds of therapeutic success.
Absorption
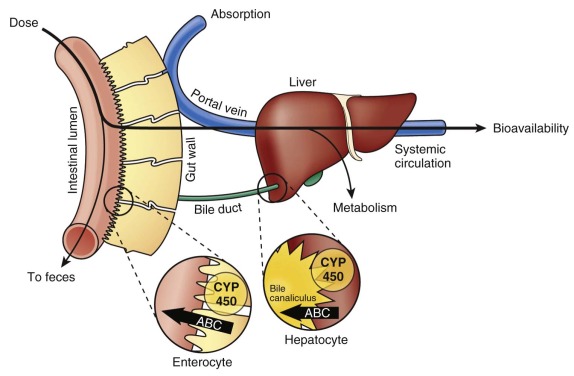
Medications are typically administered via the gastrointestinal tract (mainly orally), intravenously (IV), intramuscularly, and via the skin (subcutaneous or transdermal). Orally administered agents must surmount multiple barriers to reach the systemic circulation ( Figs. 46-2 and 46-3 ). Gastric pH, gastrointestinal motility, transporter-mediated uptake and efflux, and metabolism before a drug enters the systemic circulation play important roles in the absorption of many drugs. Although chemotherapy is rarely administered intramuscularly or transdermally, the lack of muscle and fatty tissue in newborns and infants and their thin skin would cause erratic absorption and increase the risk of toxicity.
Gastric pH is neutral during the first few weeks of life and does not approximate adult values until the age of 2 years, which may affect the bioavailability of compounds. Higher pH delays the absorption of weak acids and increases the absorption of weak bases. Methotrexate absorption, for example, is significantly reduced by coadministration with milk, which may further increase gastric pH. The solubility of tyrosine kinase inhibitors (e.g., dasatinib and nilotinib) is also dependent on pH. Further, pH-dependent drug degradation in the stomach affects the quantity of intact drug that reaches the small intestine. Absorption of lipophilic drugs requires their micellar solubility, which is dependent on biliary acids, whose secretion in turn is affected by food intake. Bile acid secretion is low at birth and increases to the adult level at approximately 6 months.
Gastric emptying time varies with gestational age and is typically longer in premature infants and neonates than in older children. Low gastrointestinal motility persists until 6 to 8 months of age and may cause delayed absorption or increased absorption because of greater contact time. In addition, drugs may be administered in altered forms to small children. For example, tablets may be crushed and added to food or slurries/suspensions and sometimes may be given via nasogastric tube, which may alter their rate and extent of absorption.
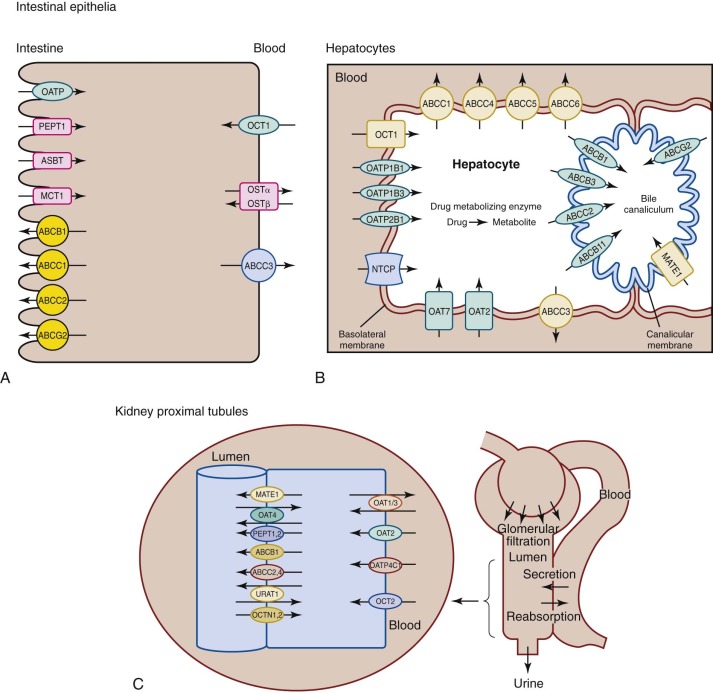
Transporter proteins are involved in drug uptake and movement across the intestinal epithelium ( Fig. 46-3 ). Intestinal epithelial cells contain several uptake transporters, including one or more members of the organic anion-transporting polypeptide (OATP) family, peptide transporter (PEPT)–1 (PEPT1; solute carrier [SLC]–15A1 [SLC15A1]), ileal apical sodium/bile acid cotransporter (SLC10A2), and monocarboxylic acid transporter 1 (SLC16A1) in their apical (luminal) membranes ( Fig. 46-4, A ). Adenosine triphosphate–binding cassette (ABC) transporters such as ABCB1 (P-glycoprotein, multidrug resistance protein [MDR]-1), ABCC1 (multidrug resistance-associated protein [MRP]–1), ABCC2 (MRP2), and ABCG2 (breast cancer resistance protein [BCRP]) are located on the apical membranes of intestinal epithelial cells, where they secrete substrates back into the intestinal lumen, thereby limiting intestinal absorption and decreasing bioavailability. The basolateral membranes of intestinal epithelial cells contain organic cation transporter–1 (OCT1; SLC22A1), heteromeric organic solute transporter α-β, and ABCC3 (MRP3). Some drugs are eliminated by hepatocytes into the bile before they are metabolized ( Figs. 46-3 and 46-4, B ). Phase I (e.g., cytochrome P [CYP]–450) and phase II (e.g., glutathione S -transferase) drug-metabolizing enzymes in the gastrointestinal tract can metabolize drugs before they enter the systemic circulation (see Fig. 46-3 ). Although the data are limited, some studies have shown that developmental changes in at least some of these enzyme activities and transporter proteins can affect drug absorption.
Volume of Distribution
The volume of distribution (Vd) of a systemically absorbed drug is the extent to which the drug penetrates extravascular tissues (see Fig. 46-2 and Table 46-2 ). Vd is affected by body composition (water and fat) and plasma proteins. Changes in body composition between birth and adolescence can alter pharmacokinetic parameters. The proportions of body water compartments, particularly extracellular fluid volume, change dramatically between birth and adulthood; extracellular fluid volume represents 50% of body weight in premature infants, 35% in infants 4 to 6 months old, and 20% in adolescents and adults. Thus polar drugs, which are distributed primarily in body water, will have a larger volume of distribution in infants than in older children and adults. The net result of an isolated increase in Vd is a lower peak concentration and prolonged terminal half-life, assuming that drug clearance remains unchanged.
Newborns have less skeletal muscle mass and subcutaneous fat than do older infants. Fat components gradually increase after birth, but they differ in boys and girls after the onset of puberty. During adolescence, boys actually lose body fat (reaching a mean of 12% of body weight), whereas girls gain body fat (reaching a mean of 25% of body weight). Therefore gender-related differences in Vd and clearance of lipophilic drugs may be more prominent during adolescence than in preadolescent children or older adults.
Drug distribution may also be affected by plasma proteins. Theoretically, drug displacement from blood components by lower protein binding increases the apparent distribution volume. Although the resulting increase in the free drug fraction may make the drug more available to its target, it also enhances metabolic and renal elimination. Protein binding may be reduced because of persistence of fetal albumin or by low plasma protein content, particularly albumin, α1-acid glycoprotein, and gamma globulin. Although the protein binding of acidic drugs may reach adult levels between 1 and 2 years of age, adult gamma-globulin levels are not reached until age 7 to 12 years.
One unique aspect of infancy is the immaturity of specific organs. For example, the myelin content of the brain is lower in newborns. Because of incomplete maturation of the blood-brain barrier (which reaches adult level by age 3 years), membrane permeability is greater in the infant brain, and the brain-to-plasma ratio of some drugs has been shown to vary with age.
Hepatic Metabolism
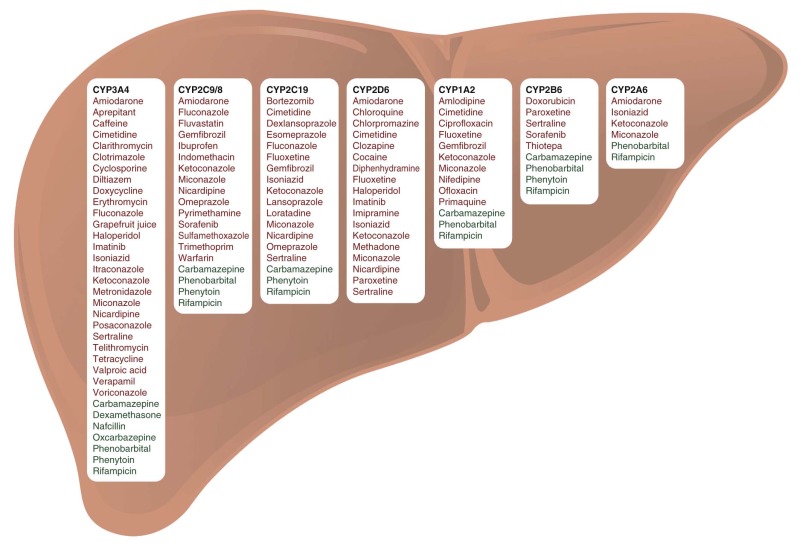
The liver plays an important role in drug metabolism (see Fig. 46-2 ). Drug-metabolizing enzymes are divided into two classes: phase I (oxidation, reduction, hydrolysis, or demethylation) and phase II (conjugation or coupling of endogenous molecules, including glucuronide, sulfate, acetate, amino acid, methyl, and glutathione moieties, to the parent drug or a phase I metabolite; Table 46-3 ). Immature organ systems in infants and young children can alter the disposition of many classes of drugs, including antibiotics, anticonvulsants, and antineoplastic agents. Phase I enzymes (e.g., CYP450 microsomal enzymes) are highly expressed in the liver. They can diminish the pharmacologic activity of a drug but can also convert a prodrug to its biologically active form (e.g., cyclophosphamide and codeine). CYP3A4 is the phase I enzyme involved in the metabolism of the largest number of drugs, followed in approximate order by CYP2D6, CYP2C8/9/19, CYP1A2, CYP2B6, CYP2A6, and CYP2E1. CYP3A7 is highly expressed in the fetus and then decreases rapidly after birth. The metabolic activity of other CYP450 enzymes (e.g., CYP3A4, CYP2D6, and CYP2C9) is low in neonates but increases dramatically between age 2 weeks and 3 years. Clearance rates by CYP450 enzymes rise from 20% of that in adults to two to six times that in adults between infancy and adolescence, and then they decline gradually to adult values during puberty. Low hydrolysis rates have been reported for drugs such as procaine in premature infants and neonates, but normal activity is measurable by 12 months. Phase II glucuronidation is almost exclusively a detoxification mechanism that inactivates a compound and facilitates its excretion by enhancing its hydrophilicity and its recognition by biliary canicular efflux proteins. The activity glucuronidation enzyme rises to 30% of adult activity by 3 months of age and then reaches adult levels between 6 and 12 months of age. Esterase activity, which is important for the metabolisms of agents such as irinotecan, gradually increases during the first year of life.
| Enzymes | Reactions |
|---|---|
| Phase I Enzymes | |
| Cytochrome P450s | C and O oxidation, dealkylation, others |
| Flavin-containing monooxygenases | N, S, and P oxidation |
| Epoxide hydrolases | Hydrolysis of epoxides |
| Phase II Enzymes | |
| UDP-glucuronosyltransferases | Addition of glucuronic acid |
| Glutathione- S -transferases | Addition of glutathione |
| Sulfotransferases | Addition of sulfate |
| N -acetyltransferases | Addition of acetyl group |
| Methyltransferases | Addition of methyl group |
| Other Enzymes | |
| Alcohol dehydrogenases | Reduction of alcohols |
| Aldehyde dehydrogenases | Reduction of aldehydes |
| NADPH-quinone oxidoreductase | Reduction of quinones |
Renal and Hepatic Excretion
The kidney is predominantly responsible for excretion of drugs and their metabolites (see Fig. 46-2 ). The immature kidney has a low glomerular filtration rate (GFR). In full-term infants, the GFR is 40 mL/min/1.73 m 2 and varies substantially among individuals. The GFR gradually increases, reaching adult values between age 6 and 12 months. Tubular function and passive resorption may also be significantly lower in the neonate. Because tubular resorption matures more slowly than does glomerular filtration, toddlers may have remarkably high clearance rates for compounds that undergo tubular resorption in older children.
GFR, as assessed by technetium-99m diethylenetriamine pentaacetic acid filtration, was related to body surface area (BSA) in children 2 months to 17 years of age, but GFR was not related to age when normalized to BSA. Premature and term infants require dose adjustments based on GFR measurement or estimation for drugs eliminated primarily by glomerular filtration, such as carboplatin. Formulas are available for estimation of creatinine clearance in infants and older children on the basis of serum creatinine and height or on another endogenous surrogate marker of GFR, such as cystatin C.
Many drugs are excreted in bile as the parent compound or as a metabolite. Biliary excretion favors compounds that have a molecular weight greater than 300 and contain both polar and lipophilic groups. Conjugation, particularly with glucuronate, increases biliary excretion. After the bile empties into the intestine, a fraction of the drug may be reabsorbed and eventually return to the liver (enterohepatic cycling). The remainder is excreted into the feces. Biliary excretion is low at birth and reaches adult levels at approximately 6 months of age.
Many SLC family transporters and ABC family transporters have been identified; these transporters mainly facilitate cellular drug uptake and elimination in kidney and liver (see Fig. 46-4, B and C ). The apical (luminal) membranes of renal proximal tubules contain organic anion transporter (OAT)–4 (OAT4, SLC22A11), urate transporter–1 (SCL22A12), PEPT1 (SLC15A1) and PEPT2 (SLC15A2), ABCC2 and ABCC4 (MRP4), multidrug and toxin extrusion (MATE) protein–1 (MATE1, SLC47A1), ABCB1, organic cation/ergothioneine transporter (OCTN1; SLC22A4), and organic cation/carnitine transporter (OCTN2; SLC22A5). The basolateral membranes of the proximal tubules contain the uptake transporters OATP4C1 (SLCO4C1); OCT2; and OAT1, OAT2 (SLC22A7), and OAT3 (SLC22A8).
Uptake transporters in the hepatocyte basolateral (sinusoidal) membrane include the sodium/taurocholate cotransporting peptide (SLC10A1), three members of the OATP family (OATP1B1 [SLCO1B1], OATP1B3 [SLCO1B3], and OATP2B1 [SLCO2B1]), OAT2 and OAT7 (SLC22A9), and OCT1. Efflux pumps in the hepatocyte basolateral membrane include ABCC1 (MRP1), ABCC3, ABCC4, ABCC5 (MRP5), and ABCC6 (MRP6). Apical (canalicular) efflux pumps of the hepatocyte comprise ABCB1, ABCC2, ABCB3 (transporter associated with antigen processing 2 [TAP2]), ABCB11 (bile salt export pump [BSEP] or sister of P-glycoprotein [SPGP]), ABCG2, and MATE1.
ABCB1 activity in lymphocytes peaks at birth, decreases between ages 0 and 6 months, and stabilizes at the adult level between the ages of 6 months and 2 years. Developmental changes in the functions of other transporters have not been fully defined, but many are under investigation.
Influence of Underlying Disease on Metabolism
Pathophysiologic changes associated with specific pediatric malignancies may alter drug disposition. For example, the clearance of antipyrine and lorazepam was observed to increase between diagnosis and the end of remission induction therapy in children with ALL. The clearance of unbound teniposide is lower in children with relapsed ALL than during first remission. Because leukemic infiltration of the liver is common at the time of diagnosis of ALL, drugs metabolized by the liver may have reduced clearance, as has been documented in preclinical models.
In mouse models, certain tumors elicited an acute-phase response that coincided with downregulation of human hepatic CYP3A4 and of the mouse orthologue Cyp3a11. The reduction of murine hepatic Cyp3a gene expression in tumor-bearing mice resulted in decreased Cyp3a protein expression and consequently a significant reduction of Cyp3a-mediated metabolism of midazolam. These findings support the possibility that tumor-derived inflammation may alter the pharmacokinetic and pharmacodynamic properties of CYP3A4 substrates, leading to reduced drug metabolism in humans.
Unique Drug Effects in Pediatric Patients
Experience has shown that children often tolerate the conventional toxicities of chemotherapy better than adults, and that children (with the exception of neonates) experience very low regimen-related mortality. This finding may be partly explained by relatively low drug exposure caused by higher drug clearance rather than by altered pharmacodynamics. Lower exposure may explain briefer periods of neutropenia, less mucositis, and rare hepatic veno-occlusive disease. Children’s greater ability to tolerate intensive therapy may partly explain why children with ALL have a higher cure rate compared with adults.
However, the immature organ systems of the very young may be more susceptible to certain aspects of toxicity, including late cardiomyopathy, neuron developmental disorders, delayed puberty, growth plate arrest, and infertility. Children appear to be more sensitive to the cardiotoxic effects of anthracyclines (e.g., doxorubicin, daunorubicin, idarubicin, and mitoxantrone). Follow-up studies have shown that months or years after intrathecal therapy for central nervous system (CNS) leukemia, with or without cranial irradiation, children treated for ALL may experience a chronic demyelinating encephalopathy and/or cognitive late effects. Further, the latent effects of chemotherapy on fertility appear to vary with the developmental stage of the patient at the time of treatment. Treatment of leukemia and lymphoma in prepubertal boys, for example, does not appear to produce sustained testicular damage unless large cumulative doses of alkylating agents (e.g., cyclophosphamide >6 g/m 2 ) are used. Treatment during puberty is more likely to permanently damage the germinal epithelium, although sexual maturation may proceed on a normal schedule. Most prepubertal girls treated for leukemia with antimetabolites achieve menarche and progress through puberty. Ovarian failure is associated mainly with alkylating agents and gonadal radiotherapy and is directly related to cumulative dose and age at exposure. Nearly 30% of survivors treated with alkylating agents and abdominal-pelvic irradiation experienced nonsurgical primary ovarian failure (defined as cessation of menses before age 40 years). Procarbazine exposure at any age, cyclophosphamide exposure from age 13 to 20 years, and ovarian radiation doses greater than 10 Gy were independent risk factors for acute ovarian failure (defined as loss of ovarian function within 5 years after diagnosis). Most girls treated for brain tumors with procarbazine and/or nitrosourea and with neuraxis radiation showed evidence of ovarian hormonal failure.
Drug Interactions
Combination Chemotherapy
The vast majority of pharmacologic studies of anticancer agents have modeled the effects of a single drug. However, as a consequence of somatic mutations and/or selection of genetically resistant clones, tumor cytotoxicity tends to diminish over subsequent courses of single-agent treatment. Therefore cancer chemotherapy is most frequently given as a combination of drugs that individually have shown activity against the specific cancer to be treated but that have different pharmacologic mechanisms of action. The selection of agents that exert maximal anticancer effects and minimal adverse effects requires a clear understanding of the biochemical, molecular, and pharmacokinetic mechanisms of action and of individual and overlapping toxicities of the agents. The treatment for ALL provides an example of excellence. Most of the drugs used were developed before 1970. However, their dosage and schedule of administration have been optimized on the basis of leukemic-cell biologic features, response to therapy (e.g., minimal residual disease), and patient pharmacodynamic and pharmacogenomic findings. The 5-year survival rate for pediatric ALL has been approximately 90% in recent trials.
Optimal combination chemotherapy theoretically should meet the following criteria:
- •
Drugs that show single-agent activity should be selected. Because of primary resistance, rates of complete response to a single agent rarely exceed 20%.
- •
Drugs with different mechanisms of action should be combined. The various classes of anticancer agents have different cellular targets. The use of multiple agents with different mechanisms of action enables independent killing of cells by each agent. Cells resistant to one agent may be sensitive to another drug or drugs in the regimen. Known patterns of cross-resistance must be taken into consideration.
- •
Drugs with different mechanisms of resistance should be combined. Resistance to many agents may be the result of mutations selected by those agents. However, a single mutational change can also cause resistance to multiple drugs. The number of known mechanisms of resistance is continuously increasing and is partly drug dependent. Because drug-resistant mutants may be present at the time of clinical diagnosis, the earliest possible use of non–cross-resistant drugs is recommended to avoid the selection of double mutants. Adequate cytotoxic doses of drugs must be administered as frequently as possible to achieve maximal killing of sensitive and moderately resistant cells.
- •
If possible, drugs with different dose-limiting toxicities should be combined, because it is more likely that drugs with nonoverlapping toxicities can be used at the full dosage, optimizing their potential effectiveness.
As noted, multidrug therapy can give rise to clinically important drug-drug interactions, which typically occur when the pharmacokinetic behavior of one drug is altered by the other ( Fig. 46-5 and Table 46-1 ). These interactions are important in the design of drug combinations, because diminished therapeutic efficacy or increased toxicity of one or more of the administered agents can occur. Combinations of drugs may also show pharmacodynamic interactions that cannot be explained by altered pharmacokinetic profiles. Some of these interactions are at the cellular level or are related to the cell cycle. Interactions can be classified as synergistic (i.e., the effect of the combination is greater than the sum of the individual effects), additive (the effect of two drugs is the sum of the effects of each), or antagonistic (the effect of two drugs is less than the sum of their individual effects). Provided that the drugs used are active against a particular disease, knowledge of cellular kinetics can be used to consider the use of non–cell cycle phase-specific agents (e.g., alkylating agents, platinum agents, and anthracyclines), first to reduce tumor bulk and second to recruit slowly dividing cells into active deoxyribonucleic acid (DNA) synthesis. After the latter is achieved, treatment can be continued with cell-cycle phase-specific agents (e.g., methotrexate or fluoropyrimidines), which affect cells mainly during DNA synthesis. Similarly, repeated courses of S-phase–specific drugs, such as cytarabine and methotrexate, are most effective if administered during the rapid rebound recovery of DNA synthesis that follows suppression of DNA synthesis in the previous course. If pharmacokinetic and pharmacodynamic interactions exist, the drug doses and sequence of administration that allow safe administration of combination chemotherapy are typically defined during early clinical trials (i.e., phase I and phase II studies).
Coadministration of Non-Anticancer Drugs
Many prescription and over-the-counter medications are used during chemotherapy to improve quality of life or to treat other diseases, but they may interact with anticancer agents, thus altering their pharmacokinetic characteristics, clinical effectiveness, and/or toxicities. More than 100,000 deaths per year in the United States alone are attributed to drug-drug interactions, placing these interactions between the fourth and sixth leading cause of death. Clearly, all aspects of pharmacokinetics (i.e., absorption, distribution, metabolism, and excretion) may be affected when a drug is given in combination with another drug. The generally narrow therapeutic index of cytotoxic chemotherapy means that drug-drug interactions would predispose some persons to excessive toxicity or inadequate efficacy.
Pharmacokinetic Interactions Affecting Drug Absorption
Because many chemotherapeutic agents are given via an IV line, factors that affect absorption have little effect on their pharmacokinetics. However, absorption plays a crucial role in the bioavailability of orally administered drugs; absorption is contingent on adequate intestinal uptake and circumvention of intestinal and subsequent hepatic metabolism of the drug (see Figs. 46-2 and 46-3 ). Food intake can delay gastric emptying, raise intestinal pH, increase hepatic blood flow, and slow gastrointestinal transit, which may significantly affect the pharmacokinetic profile of orally administered medications. Mercaptopurine is metabolized by xanthine oxidase into inactive metabolites. Ingestion of foods that contain xanthine oxidase, such as dairy products, can reduce the effectiveness of mercaptopurine. Therefore patients are instructed to take mercaptopurine on an empty stomach, preferably at night (2 hours after food intake). Proton-pump inhibitors and antacids increase gastric pH and decrease the solubility and absorption of tyrosine kinase inhibitors (with the exception of imatinib).
Another important mechanism of interaction with orally administered anticancer agents is their affinity for ABC transporters (e.g., ABCB1, ABCC1, ABCC2, and ABCG2), which are located on the apical membranes of intestinal epithelial cells and secrete substrates from the epithelial cell back into the intestinal lumen, thereby limiting intestinal absorption and bioavailability (see Figs. 46-3 and 46-4, A ). Some chemotherapeutic agents are substrates of these transporters, the activity of which may be inhibited by other medications, foods, and herbal products. In addition, extensive metabolism of anticancer drugs in the gut wall and/or the liver (e.g., by CYP450 enzymes) during the first pass can keep them out of the systemic circulation and is another potential drug-drug interaction mechanism (see Fig. 46-3 ). An ideal chemotherapeutic drug would have an adequate absolute bioavailability, as shown by little drug-drug interaction or interpatient/intrapatient variability in absorption. However, the most commonly used oral agents, including mercaptopurine, methotrexate, etoposide, and cyclophosphamide, do not meet these criteria.
Pharmacokinetic Interactions Affecting Drug Distribution
Chemotherapeutic agents can bind to several blood components, including albumin, lipoproteins, immunoglobulins, and α1-acid glycoprotein, and the increased free-drug fraction caused via displacement by another drug may make the displaced drug more available to the target and enhance metabolism and elimination. Highly protein-bound drugs, such as warfarin, may interact with protein-bound cytotoxic drugs such as etoposide and paclitaxel. The hematologic toxicity of etoposide is more closely related to systemic exposure to the unbound drug than to the total drug. However, the therapeutic implications of displacement of many anticancer drugs from their protein-bound state are as yet undefined.
Pharmacokinetic Interactions Affecting Drug Metabolism
Most important drug-drug interactions involve altered liver metabolism—for example, altered expression or functionality of CYP450 isoenzymes, such as CYP3A4, 2C9/8, 2D6, and 2C19 isoforms, which oxidize anticancer drugs to more polar and usually inactive metabolites (see Table 46-1 ). Induction of CYP450 activity, which increases a drug’s metabolic rate, may reduce plasma drug concentration and thus reduce the therapeutic effect. For example, anticonvulsant drugs (such as phenytoin, phenobarbital, and carbamazepine) induce drug-metabolizing CYP450 enzymes and thereby increase the clearance of various anticancer agents (e.g., anthracyclines, epipodophyllotoxins, and vinca alkaloids) in children (see Table 46-1 and Fig. 46-5 ). Enzyme-inducing anticonvulsants have been shown to be associated with lower event-free survival, hematologic relapse, and CNS relapse in persons with ALL. Conversely, suppression (inhibition) of CYP450 activity may increase plasma drug concentration, causing greater toxicity commensurate with overdose. Valproic acid, azole antifungals (e.g., fluconazole, voriconazole, and posaconazole), the antiemetic aprepitant, and macrolide antibiotics (e.g., clarithromycin, erythromycin, and azithromycin) should be avoided or prescribed with caution when CYP450-metabolized chemotherapeutic agents are given concomitantly. Although drugs are typically metabolized to their inactive forms, prodrugs such as cyclophosphamide are metabolized to their active forms by CYP450 enzymes and have the opposite consequences of metabolic inhibition or activation.
Pharmacokinetic Interactions Affecting Drug Excretion
ABC transporters such as ABCB1, ABCC1/2, and ABCG2 are involved in drug elimination. ABCB1 in the brush-border membrane of the proximal renal tubule plays an important role in renal elimination of chemotherapeutic agents by active secretion into the urine (see Fig. 46-4, C ). Inhibition of ABCB1 can result in increased systemic exposure and tissue distribution of drugs that are ABCB1 substrates (see Table 46-1 ), whereas induction of ABCB1 leads to decreased systemic exposure. ABCB1 is also expressed in canalicular membranes of the liver; it has a role in elimination of drugs into bile, from which they can be reabsorbed in the intestine or eliminated in the feces (see Fig. 46-4, B ). Excretion of irinotecan and its metabolites is mediated by several ABC transporters (e.g., ABCB1 and ABCG2). Substrates and/or inhibitors of these transporters (e.g., cyclosporine and gefitinib) may interfere with renal and/or biliary excretion of irinotecan and its metabolite, SN-38, and increase the plasma concentration and toxicity. Organic anion transporters (e.g., OAT1, OAT3, and OAT4) are highly expressed on the renal proximal tubules and are involved in the renal elimination of methotrexate. Nonsteroidal antiinflammatory drugs (e.g., salicylate, ibuprofen, and indomethacin), probenecid, and penicillin can inhibit these OATs and delay renal excretion of methotrexate, leading to severe and even life-threatening drug interactions, including bone-marrow suppression and acute renal failure.
Coadministration of Complementary and Alternative Medicine
In recent years, interest in complementary and alternative medicine (CAM) has grown rapidly in the industrialized world. No compelling clinical trial data support the use of CAM in children with cancer, and the potential interaction of CAM agents with conventional, possibly curative, treatment may be harmful. Families are more likely to use CAM for their child if a parent uses CAM, if the child has a poor prognosis, or if the parents are older and better educated. Surveys within the past decade have estimated that CAM is used in 31% to 84% of pediatric oncology cases, and in many cases the treating clinician is unaware of the patient’s use of CAM. In addition to nonherbal therapies (e.g., hypnosis, guided imaginary, massage, and acupuncture), many children use herbal treatments, such as St. John’s wort ( Hypericum perforatum ), garlic, and ginseng ( Panax ginseng ), in combination with allopathic therapies. The risk of herb-drug interactions is a growing concern, and the need is increasing to understand possible adverse interactions. This concern is of particular relevance to pediatric oncology, because CAM use is more common in this group than in the general population.
During the past decade, a wealth of in vitro and in vivo evidence has shown that many herbal preparations interact extensively with drug-metabolizing enzymes and drug transporters. A number of clinically important interactions are now recognized, although causal relationships have not consistently been established and confirmatory studies in children are lacking. Most of the observed interactions suggest that the herbs inhibit or induce several isoforms of the CYP family, and concurrent use of some herbs with chemotherapy is likely to have serious clinical and toxicologic implications. An additional consideration for cancer chemotherapy is that herb-mediated induction or inhibition of phase I and II metabolic enzymes and ABC/SLC transporters may alter response. Therefore rigorous testing is urgently required to identify the pharmacokinetic interactions of anticancer drugs with widely used herbs. Drug-drug interaction potential has been investigated for only a limited number of herbs. For example, St. John’s wort, melatonin, and whey protein are CYP450 inducers, and curcumin, goldenseal (Hydrastis canadensis) , and grapefruit juice (Citrus paradise) are strong inhibitors. Milk thistle (Silybum marianum) , Echinacea (Echinacea purpurea) , saw palmetto, Ginkgo biloba , valerian, and green tea have showed no significant interactions. Because of the widespread use of herbal medicines in the United States, physicians should include it in their routine drug histories and review potential hazards with individual patients. Detailed information can be obtained from the following websites :
- •
National Center for Complementary and Alternative Medicine, National Institutes of Health: http://nccam.nih.gov.easyaccess2.lib.cuhk.edu.hk
- •
Office of Cancer Complementary and Alternative Medicine, National Cancer Institute, National Institutes of Health: http://cam.cancer.gov/
- •
PDQ Cancer Information Summaries: Complementary and Alternative Medicine, National Cancer Institute, National Institutes of Health: http://www.cancer.gov/cancertopics/pdq/cam
- •
Integrative Therapies Program for Children with Cancer, Columbia University Medical Center: http://integrativetherapies.columbia.edu/
- •
About Herbs, Botanicals and Other Products, Memorial Sloan Kettering Cancer Center: http://www.mskcc.org/mskcc/html/11570.cfm
- •
Complementary/Integrative Medicine, MD Anderson Cancer Center: http://www.mdanderson.org/departments/cimer/
- •
Natural Standard: http://www.naturalstandard.com/
Pharmacogenetics and Pharmacogenomics of Chemotherapeutic Agents in Children
Pharmacogenetics/pharmacogenomics investigates the influence of human genomic variation in drug response. Inherited (germline) or acquired (somatic) variation in genes encoding drug-metabolizing enzymes, drug receptors, drug transporters, and drug targets have all been shown to influence anticancer drug response in humans and they may also influence drug toxicity.
A single nucleotide polymorphism (SNP) is defined as a single base change in a DNA sequence. Unless it occurs in at least 1% of the population, it is considered a variant or a mutation. Accumulation of mutations during evolution, in concert with random selection, has made SNPs the most common form (about 90%) of genetic variation in the human genome.
The advent of pharmacogenomics was characterized by the passage from a candidate gene approach to a genome-wide approach. This advance was initially made possible by the international HapMap project, which included 270 samples from three geographic areas of genetic descent—African descent (Yoruban population); European descent (Center d’Etude du Polymorphisme Humain population), and Asian descent (Japanese and Han Chinese population)—and more than 2 million publicly available SNPs. Subsequently, the “1000 Genomes Project” has provided the most detailed catalogue of human genetic variation. The recent development of next-generation sequencing technologies has allowed better coverage of all genetic variants.
Advances in treatment have greatly improved the survival rates of children with cancer. However, the efficacy of therapy, as well as its short- and long-term adverse effects, can vary among patients, even when their chemotherapeutic or radiologic treatment is identical. Thus it is very important to identify inherited (germline) genomic variations that can alter the absorption, distribution, metabolism, and/or excretion of chemotherapeutic agents, thereby altering their effects and toxicities ( Table 46-4 ).
| Agent | Genes Involved | Effects |
|---|---|---|
| Thiopurines | TPMT*3A (460 G>A and 719 A>G), TPMT*3C (719 A>G), and TPMT*2 (238G>C) | Higher incidence of hematopoietic cell toxicities, secondary malignancies in ALL |
| 6-mercaptopurine | PACSIN2 (rs2413739, T allele) | Lower TPMT activity and higher incidence of severe gastrointestinal toxicity in ALL |
| 6-thioguanine | ITPA (rs41320251, C>A) | Higher methylmercaptopurine nucleotides and higher incidence of febrile neutropenia when mercaptopurine dose was adjusted with TPMT genotype in ALL |
| Methotrexate | SLC19A1 (G80A, AA or AG vs. GG) | Worse EFS and higher gastrointestinal toxicities in ALL |
| SLC19A1 AA80 and positive GSTM1 | Higher incidence of hepatotoxicity in ALL | |
| DHFR (G308A, AA or AG vs. GG) and DHFR *1b haplotype | Worse EFS in ALL | |
| TYMS (3/3 vs. 2/3 or 2/2) | Increased risk of hematologic relapse in ALL | |
| MTHFR (C677T, TT vs. CC) | Increased risk of relapse in ALL | |
| MTHFR (T677A1298 haplotype) | Lower EFS in ALL, especially in the presence of TYMS (3/3) | |
| MTHFD1 (A1958 variant, AA or GA vs. GG) | Lower EFS in ALL, especially in the presence of TYMS (3/3) | |
| SLCO1B1 (rs11045879 [T allele], rs4149081 [G allele], and rs4149056 [TT]) | Higher methotrexate clearance and higher incidence of gastrointestinal toxicity in ALL | |
| Glucocorticoids Prednisone Prednisolone Dexamethasone | NR3C1 (-627 AA vs. AG or GG, intron 2+646 CG or GG vs. CC, and 9b TT vs. CC or TC) | Worse EFS in ALL |
| NR3C1 (1088 AG vs. AA) | Increased risk of hematologic relapse in ALL patients with GSTM1 null genotype | |
| GSTT1 (*0/0 vs. *A/A) | Better early response to prednisone in ALL | |
| GSTP1 (codon105 Val/Val or Val/Ile vs. Ile/Ile, codon 114 Ala/Ala vs. Ala/Val or Val/Val) | Increased central nervous system relapse in ALL | |
| GSTM1 (null vs. normal) | Severe infection in ALL | |
| SNPs in CNTNAP2, LEPR, CRHR1, NTAN1, SLC12A3, ALPL, BGLAP, and APOB | Association with hypertension during remission-induction therapy in ALL | |
| ACP1 (rs 12714403 AA or AG vs. GG) | Higher incidence of symptomatic osteonecrosis in ALL | |
| TYMS (2/2 vs 3/3 or 2/3) and VDR (rs2228570, CC vs. TT or CT) | Higher incidence of osteonecrosis in ALL | |
| PAI-1 (rs6092, AA or GA vs. GG) | Higher incidence of osteonecrosis in ALL | |
| Asparaginase | ||
| Escherichia coli asparaginase | ATF5 T1562C (TT or CT vs. CC) | Inferior EFS when treated with Escherichia coli asparaginase in ALL |
| PEG-asparaginase | SNPs in ADSL, DARS, ASS1, DARS2, NARS2, ADSSL1 | Association with in vitro asparaginase sensitivity in ALL |
| Erwinia asparaginase | GRIA1 (rs4958381, A allele vs. G allele) | Higher incidence of asparaginase hypersensitivity in ALL |
| Vincristine | VDR intron 8 (AA or AG vs. GG), CYP3A5*3 (AA or AG vs. GG) | Increased peripheral neuropathy in ALL |
| PMP22 duplication (Charcot-Marie-Tooth disease) | Increased peripheral neuropathy in ALL | |
| Anthracycline | ||
| Daunorubicin | CBR3 V244M (GG vs. GA or AA) | Increased cardiomyopathy, especially with anthracycline dose at 1-250 mg/m 2 |
| Doxorubicin | SLC28A3 (rs7853758, L461L) | Decreased risk of cardiotoxicity |
| Mitoxantrone | ABCC1 (rs3743527TT, combination of rs3743527TT-rs246221TC/TT) | Increased cardiotoxicity in ALL |
| Idarubicin | CAT (rs10836235; c.66 + 78C > T, CC vs. TC) | Increased cardiotoxicity in ALL |
| Cytarabine | SLC29A1 haplotype with −1345C>G, −1050G>A, or −706G>C | Associated with high hENT1 expression |
| DCK −360G allele (GG or CG vs. CC) | Increased risk of mucositis after low-dose cytarabine in ALL | |
| CDA A79C (CC vs. AA or AC) | Higher postinduction treatment-related mortality in AML | |
| CDA G208A (GA or AA vs. GG) | Lower activity of CDA and increased sensitivity to cytarabine in ALL, AML, and NHL | |
| Etoposide | ABCB1 exon 26 (3435C>T, CC or CT or TT) | Higher clearance in patients with ALL who have prednisone pretreatment |
| CYP3A5*3 (AA vs. AG or GG) and GSTP1 (313 A>G, AA vs. AG or GG) | Lower clearance in black patients with ALL who have prednisone pretreatment | |
| VDR intron 8 (GG vs. AG) and VDR Fok 1 (CC vs. CT or TT) | Higher clearance in black patients with ALL who do not have prednisone pretreatment | |
| UGT1A1 (7/7 vs. 6/7 or 6/6) | Lower clearance in black patients with ALL who do not have prednisone pretreatment | |
| SNPs in AGPAT2, IL1B, and WNT5B | Higher etoposide catechol AUC in black and white patients with ALL | |
| SNPs in UVRAG, SEMA5A, SLC7A6, and PRMT7 | Association with etoposide cytotoxicity | |
MDM2 309T>G | Association with etoposide cytotoxicity Decreased sensitivity to etoposide | |
| Cyclophosphamide | CYP2B6*4 (AG or GG vs. AA), recipient | Higher incidence of oral mucositis in patients with leukemia who received MSD HSCT |
| CYP2B6*2 (CT or TT vs. CC), recipient | Higher incidence of hemorrhagic cystitis in patients with leukemia who received MSD HSCT | |
| CYP2B6*6 (GG vs. GT or TT), donor | Higher incidence of SOS in patients with leukemia who received MSD HSCT | |
| CYP2B6 rs3211371, R487C (Arginine), recipient | Reduced toxicity <100 days after allogeneic HSCT | |
| SOD2 rs4880, V16A (Valine), recipient | Higher overall toxicity after allogeneic HSCT | |
| Platinum compounds Cisplatin Carboplatin Oxaliplatin | ERCC2 Lys751Gln G allele (TG or GG vs. TT) | Poor tumor response and shorter EFS in patients with osteosarcoma treated with cisplatin |
| GSTM3 (*B allele vs. *A allele) | Protective effect for cisplatin-induced ototoxicity in solid tumor and CNS malignancies | |
| GSTP1 ( 105 Val/ 105 Val vs. 105 Ile/ 105 Ile or 105 Ile/ 105 Val) | Protective effects in long-term cisplatin-induced hearing impairment in testicular cancer | |
| GSTM1 and GSTT1 polymorphisms (≥1 null type vs. non-null type) | More cognitive impairment after therapy in children with medulloblastoma | |
| TPMT (rs12201199, A allele) and COMT (rs9332377, A allele) | Increased cisplatin-induced hearing loss in patients with solid tumor and CNS malignancies | |
| Megalin (rs2075252, AG or AA vs. GG) | Increased cisplatin-induced hearing loss in patients with solid tumor and CNS malignancies | |
| Irinotecan | UGT1A1*28 (7/7 vs. 6/7 or 6/6) | No association with severe toxicity when treated with low dose and protracted schedule |
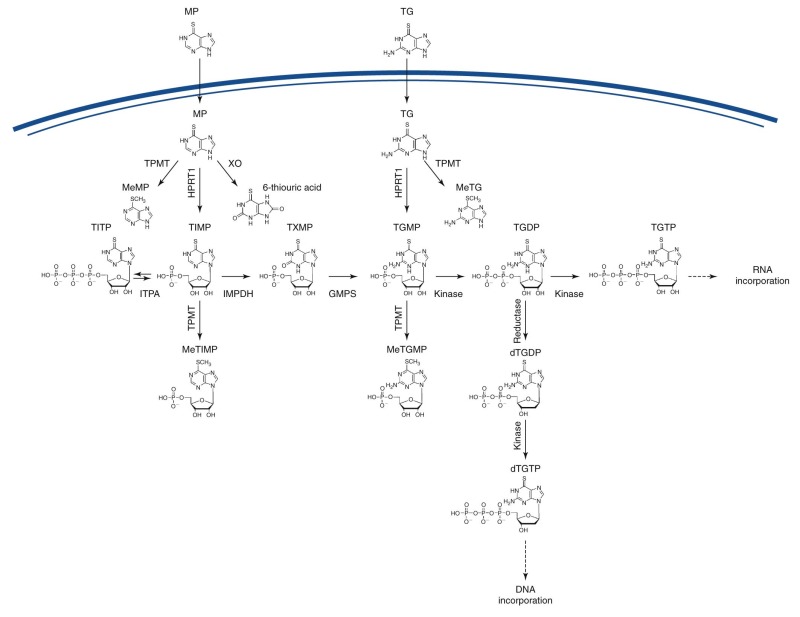
Thiopurines
Thiopurines frequently used for the treatment of childhood ALL are 6-mercaptopurine and 6-thioguanine (see Table 46-1 ). As prodrugs, they require conversion to thioguanine nucleotides (TGNs) via the purine-salvage pathway, and 6-thioguanosine 5′-triphosphate (TGTP) and deoxy-TGTP are then incorporated into ribonucleic acid (RNA) or DNA, respectively ( Fig. 46-6 ). The active DNA mismatch repair system identifies these base-pair mismatches and induces cell cycle arrest and apoptosis. An alternative protein complex that includes high mobility group box 1 and 2 proteins, heat shock cognate protein 70 (HSC70), protein disulfide isomerase (ERp60), and glyceraldehyde 3-phosphate dehydrogenase trigger apoptosis when it detects changes in DNA structure caused by incorporation of nonnatural nucleosides. Thiopurines are inactivated by methylation catalyzed by thiopurine methyltransferase (TPMT). The relation between TPMT polymorphisms and thiopurine efficacy and toxicity provides a well-established example of the potential clinical importance of pharmacogenomics ( Fig. 46-7 and Table 46-4 ). SNPs in the TPMT gene are associated with different inherited levels of TPMT enzyme activity. One in approximately 400 white persons has profound TPMT deficiency resulting from inheritance of two nonfunctional variants of TPMT , 6% to 11% have intermediate activity by having heterozygous variants of TPMT , and 89% to 94% have normal activity (homozygous wild-type TPMT ). Ethnic differences exist in the frequency of occurrence of these variant alleles and thus in the frequency of TPMT deficiency. To date more than 20 TPMT variants have been identified, but three variants— TPMT*3A (460 G>A and 719 A>G), TPMT*3C (719 A>G), and TPMT*2 (238 G>C)—account for more than 90% of inactivating alleles.