Cancer Chemotherapeutic Aents
Dennis A. Casciato
I. ALKYLATING AGENTS
A. General pharmacology of alkylating agents. Alkylating agents target DNA and are cytotoxic, mutagenic, and carcinogenic. All agents produce alkylation through the formation of intermediates.
1. Alkylating agents impair cell function by transferring alkyl groups to amino, carboxyl, sulfhydryl, or phosphate groups of biologically important molecules. Most importantly, nucleic acids (DNA and RNA) and proteins are alkylated. The number 7 (N-7) position of guanine in DNA and RNA is the most actively alkylated site; the O-6 group of guanine is alkylated by nitrosoureas. Alkylation of guanine results in abnormal nucleotide sequences, miscoding of messenger RNA, cross-linked DNA strands that cannot replicate, breakage of DNA strands, and other damage to the transcription and translation of genetic material.
2. The primary mode of action for most alkylating agents is by means of cross-linking of DNA strands. Cytotoxicity is probably a result of damage to the DNA templates rather than inactivation of DNA polymerase and other enzymes responsible for DNA synthesis. DNA strand breakage also appears to be a minor determinant of cytotoxicity.
3. Alkylating agents are cell cycle-specific but not phase-specific. The drugs kill a fixed percentage of cells at a given dose.
4. Tumor resistance to these drugs appears to be related to the capacity of cells to repair nucleic acid damage and to inactivate the drugs by conjugation with glutathione.
B. Bendamustine (Treanda)
1. Indications. Chronic lymphocytic leukemia (CLL), low grade B-cell non-Hodgkin lymphoma (NHL) that has progressed within 6 months of treatment with a rituximab-containing regimen
2. Pharmacology. Bendamustine is a bifunctional mechlorethamine derivative containing a purine-like benzimidazole ring.
a. Mechanisms. Alkylation (see Section A). The exact mechanism of action is unknown.
b. Metabolism. About 90% of the drug is excreted in the feces
3. Toxicity (adverse reactions are more common when dosing for NHL than for CLL). The drug is an irritant.
a. Dose-limiting. Hematosuppression
b. Common. Nausea, vomiting, diarrhea; fever, fatigue, headache, stomatitis, rash
c. Occasional. Anaphylactic reactions, severe skin reactions, acute renal failure; peripheral edema, tachycardia, hypotension, dizziness; myelodysplasia; dysgeusia
4. Administration. Fever, chills, pruritus, and rash commonly occur during the infusion (“infusion reactions”); consider administering an antihistamine, acetaminophen, and corticosteroids prophylactically if these reactions develop and are mild.
a. Supplied as 100 mg vial
b. Dose modification
(1) Dose modification for renal and hepatic function. Use with caution in mild renal or hepatic impairment; do not use if creatinine clearance is <40 mL/min or in moderate or severe hepatic impairment.
(2) Dose modification for grade 3 or higher toxicity: For CLL, reduce dose to 50 mg/m2 on days 1 and 2 of cycle and to 25 mg/m2 on days 1 and 2 if toxicity recurs. For NHL, reduce dose to 90 mg/m2 on days 1 and 2 of cycle and to 60 mg/m2 on days 1 and 2 if toxicity recurs.
c. Dose: For CLL, 100 mg/m2 IV over 30 minutes on days 1 and 2 of a 28-day cycle; for NHL, 120 mg/m2 IV over 60 minutes on days 1 and 2 of a 21-day cycle
d. Drug interactions. Concomitant CYP1A2 inducers or inhibitors may affect the exposure of bendamustine. Drugs that induce CYP1A2 include barbiturates, carbamazepine, phenobarbital, rifampin, and tobacco smoking. Drugs that inhibit CYP1A2 include cimetidine, ciprofloxacin, antiretrovirals, and mexiletine.
C. Busulfan (Myleran)
1. Indications. Chronic myelogenous leukemia (CML) palliation, bone marrow transplantation (high doses)
2. Pharmacology
a. Mechanism. Alkylation (see Section A)
b. Metabolism. Acts directly; catabolized to inactive products that are excreted in the urine.
3. Toxicity
a. Dose-limiting. Reversible and irreversible myelosuppression with slow recovery; blood cell counts fall for about 2 weeks after discontinuation of drug.
b. Common. Gastrointestinal (GI) upset (mild), sterility
c. Occasional. Skin hyperpigmentation, alopecia, rash; gynecomastia, cataracts, LFT abnormalities; seizures
d. Rare. Pulmonary fibrosis (“busulfan lung”; see Chapter 29, Section IV.A), retroperitoneal fibrosis, endocardial fibrosis; addisonian-like asthenia (without biochemical evidence of adrenal insufficiency); hypotension, impotence, hemorrhagic cystitis, secondary neoplasms
4. Administration
a. Supplied as 2-mg tablets
b. Dose modification. Hematologic
c. Dose. Usually 2 to 8 mg/d PO; or 0.05 mg/kg/d for CML
d. Drug interactions.
(1) Drugs that induce or inhibit CYP3A4 (see Section VI.A.)
(2) Itraconazole and metronidazole reduce busulfan metabolism resulting in increased therapeutic and toxic effects. St. John’s wort increases busulfan elimination.
D. Chlorambucil (Leukeran)
1. Indications. Chronic lymphocytic leukemia (CLL), Waldenstrom macroglobulinemia
2. Pharmacology
a. Mechanism. Alkylation (see Section I.A)
b. Metabolism. Acts directly; spontaneously hydrolyzed to inactive and active products (e.g., phenylacetic acid mustard); also is extensively metabolized by the hepatic P450 microsomal system. The drug and metabolic products are excreted in urine.
3. Toxicity. Least toxic alkylating agent
a. Dose-limiting. Myelosuppression
b. Occasional. GI upset (minimal or absent at usual doses), mild LFT abnormalities, sterility, rash
c. Rare. Rash, alopecia, fever; cachexia, pulmonary fibrosis, neurologic or ocular toxicity, cystitis; acute leukemia
4. Administration
a. Supplied as 2-mg tablets
b. Dose modification. Hematologic
c. Dose. Various dosage schedules are used. For example, 0.1 to 0.2 mg/kg/d PO for 3 to 6 weeks and then decrease dose for maintenance
d. Drug interactions. Phenobarbital, phenytoin, and other drugs that stimulate the liver’s P450 system may lead to increased production of toxic metabolites.
E. Cyclophosphamide (Cytoxan)
1. Indications. Used in a wide variety of conditions
2. Pharmacology
a. Mechanism. Alkylation (see Section I.A); also inhibits DNA synthesis. Cell cycle-nonspecific and active in all phases of the cell cycle.
b. Metabolism. Native drug is inactive and requires activation by liver P450 microsomal oxidase system to form an aldehyde that decomposes in plasma and peripheral tissues to yield acrolein and an alkylating metabolite (e.g., phosphoramide mustard). The P450 system also metabolizes metabolites to inactive compounds. Active and inactive metabolites are excreted in urine.
3. Toxicity
a. Dose-limiting
(1) Myelosuppression. Leukopenia develops 8 to 14 days after administration. Thrombocytopenia occurs but is rarely significant.
(2) Effects on urinary bladder. Degradative products are responsible for hemorrhagic cystitis, which can be prevented by maintaining a high urine output. Hemorrhagic cystitis is more common and can be severe when massive doses are used (e.g., for bone marrow transplantation); under these circumstances, the use of mesna can be preventative. Urinary bladder fibrosis with telangiectasia of the mucosa can occur (usually after long-term oral therapy) without episodes of cystitis. Bladder carcinoma has occurred.
b. Side effects
(1) Common. Alopecia, stomatitis, aspermia, amenorrhea; headache (fast onset, short duration). Nausea and vomiting are common after doses of 700 mg/m2 or more.
(2) Occasional. Skin or fingernail hyperpigmentation; metallic taste during injection; sneezing or a cold sensation in the nose after injection; abnormal LFTs, dizziness; allergy, fever
(3) Rare. Transient syndrome of inappropriate secretion of antidiuretic hormone (SIADH, especially if given with a large volume of fluid), hypothyroidism, cataracts, jaundice, pulmonary fibrosis; cardiac necrosis and acute myopericarditis (with high doses); secondary neoplasms (acute leukemia, bladder carcinoma)
4. Administration. The drug should be administered with a large volume of fluid in the morning or early afternoon to avoid cystitis.
a. Supplied as 25- or 50-mg tablets; vials contain 500 to 2,000 mg
b. Dose modification. Hematologic; may be required for hepatic or renal functional impairment
c. Dose. Cyclophosphamide is frequently employed as part of combination chemotherapy regimens. Some common doses are 0.5 to 1.5 g/m2 IV every 3 weeks or 50 to 200 mg/m2 PO for 14 days every 28 days.
d. Drug interactions. Phenobarbital, phenytoin, and other drugs that stimulate the liver’s P450 system may lead to increased production of toxic metabolites.
(1) Digoxin levels decreased with cyclophosphamide.
(2) Interacts with warfarin to prolong the prothrombin time further
(3) Interacts with succinyl choline to increase neuromuscular blockade
(4) Allopurinol increases myelosuppression with concomitant use.
(5) Cimetidine may decrease rate of active metabolite formation and inhibit antineoplastic activity.
F. Dacarbazine [dimethyl-triazeno-imidazol-carboxamide (DTIC, DIC), imidazole carboxamide]
1. Indications. Hodgkin lymphoma, malignant melanoma, sarcomas, neuroblastoma
2. Pharmacology
a. Mechanisms. Inhibits purine, RNA, and protein synthesis. Has some alkylating activity. Causes DNA methylation and direct DNA damage. Cell cycle-nonspecific.
b. Metabolism. Native drug inactive; requires activation by oxidative N-methylation by the hepatic P450 microsomal system. Excreted in urine predominantly (50% of the drug is unchanged); minor hepatobiliary and pulmonary excretion.
3. Toxicity
a. Dose-limiting. Myelosuppression; nadir blood counts occur 2 to 4 weeks after treatment
b. Common. Nausea and vomiting (often severe), anorexia; pain along the injection site
c. Occasional. Alopecia, facial flushing, photosensitivity, abnormal LFTs. Flu-like syndrome (malaise, myalgia, chills, and fever) developing 1 week after treatment and lasting several days
d. Rare. Diarrhea, stomatitis; cerebral dysfunction; hepatic veno-occlusive disease, hepatic necrosis; azotemia; anaphylaxis
4. Administration. Dacarbazine is often used in combination with chemotherapy regimens. Withdrawing blood into the drug-filled syringe before injecting the mixture reduces the pain of injection. The drug is a vesicant if injected subcutaneously.
a. Supplied as 100- and 200-mg vials
b. Dose modification. Necessary for patients with impaired bone marrow, hepatic, or renal dysfunction
c. Dose
(1) 375 mg/m2 every 15 days in ABVD regimen for Hodgkin lymphoma, or
(2) 220 mg/m2 IV daily for 3 days every 21 to 28 days
d. Drug interactions.
(1) DTIC elimination may be altered by drugs that inhibit or induce the P450 system (e.g., barbiturates, phenytoin, rifampin, ciprofloxacin, isoniazid, disulfiram).
(2) DTIC may reduce levodopa effects with concomitant use; levodopa dosage increase may be required.
G. Ifosfamide (isophosphamide, Ifex)
1. Indications. Lymphomas, sarcomas, relapsed testicular tumors, and various carcinomas
2. Pharmacology
a. Mechanisms. An alkylating agent (see Section I.A); DNA cross-linking and chain breakage. Metabolites are alkylating agents that are similar to cyclophosphamide but are not cross-resistant.
b. Metabolism. Extensively metabolized by the hepatic P450 microsomal system. Activated at a fourfold slower rate than cyclophosphamide because
of a lower affinity for the P450 system. Inactive until activated by hepatic microsomal enzymes. Like cyclophosphamide, the drug undergoes hepatic activation to an aldehyde form that decomposes in plasma and peripheral tissues to yield acrolein and its alkylating metabolite. Acrolein is highly toxic to urothelial mucosa. The chloroacetaldehyde metabolite may be responsible for much of the neurotoxic effects, particularly in patients with renal dysfunction. Drug and metabolites are excreted in urine.
of a lower affinity for the P450 system. Inactive until activated by hepatic microsomal enzymes. Like cyclophosphamide, the drug undergoes hepatic activation to an aldehyde form that decomposes in plasma and peripheral tissues to yield acrolein and its alkylating metabolite. Acrolein is highly toxic to urothelial mucosa. The chloroacetaldehyde metabolite may be responsible for much of the neurotoxic effects, particularly in patients with renal dysfunction. Drug and metabolites are excreted in urine.
3. Toxicity
a. Dose-limiting. Myelosuppression, hemorrhagic cystitis, encephalopathy
b. Common. Alopecia; anorexia, constipation, nausea, and vomiting; amenorrhea, oligospermia, and infertility
c. Neurotoxicity (especially with hepatic or renal dysfunction, hypoalbuminemia, low bicarbonate levels, or with rapid infusion): Somnolence, confusion, depression, hallucinations, dizziness, cranial nerve dysfunction, and ataxia. These effects usually resolve within 3 days of discontinuation of drug.
d. Occasional. Salivation, stomatitis, diarrhea; urticaria, hyperpigmentation, nail ridging; abnormal LFTs, phlebitis, fever; hypotension, hypertension, hypokalemia; renal tubular acidosis (at high doses); SIADH
e. Rare. Coma; renal tubular acidosis, or Fanconi-like syndrome
4. Administration. Aggressive concomitant hydration (2 to 4 L/d) and mesna are given to reduce the incidence of hemorrhagic cystitis. Monitor urine for hematuria before each dose. Use antiemetics prophylactically.
a. Supplied as 1- and 3-g prepackaged vials with mesna
b. Dose modification. Hematologic and renal dysfunction
c. Dose. 1,000 to 1,200 mg/m2 IV over 30 minutes for 3 to 5 days every 3 to 4 weeks. Various dosages and dose schedules are available (e.g., see Appendix D and Chapter 17, Sarcomas).
d. Mesna (sodium 2-mercaptoethanesulfonate, Mesnex) is an uroprotective agent when administering ifosfamide or cyclophosphamide. Alternative dose schedules for mesna, which is given in the same mg dose as the alkylating agent, are
(1) Equal doses of ifosfamide and mesna in the same bag when given as a continuous infusion
(2) Twenty percent of the mesna dose given IV bolus at the time of administration of ifosfamide and then forty percent PO at 2 hours and forty percent at 6 hours after each dose of ifosfamide/cyclophosphamide
(3) When given by IV bolus, the total dose of mesna is 60% of the ifosfamide dose. One-third of the mesna dose (20% of the ifosfamide dose) is given 15 minutes before, 4 hours after, and 8 hours after ifosfamide.
e. Drug interactions. Phenobarbital, phenytoin, and other drugs that stimulate the liver’s P450 system may lead to increased production of toxic metabolites.
(1) Digoxin levels are decreased with cyclophosphamide.
(2) Interacts with warfarin to prolong the prothrombin time further
(3) Interacts with succinyl choline to increase neuromuscular blockade
(4) Cimetidine and allopurinol increase ifosfamide toxicity.
H. Melphalan (Alkeran, phenylalanine mustard, L-PAM)
1. Indications. Multiple myeloma. The injection form is used in bone marrow transplantation. Previously used in ovarian carcinoma.
2. Pharmacology
a. Mechanism. Alkylation (see Section I.A)
b. Metabolism. Acts directly. Ninety percent of the drug is bound to plasma proteins and undergoes rapid hydrolysis in the bloodstream
to inert products. Melphalan is excreted in the urine (about 30%) as unchanged drug and metabolites, and the remainder is cleared in feces.
to inert products. Melphalan is excreted in the urine (about 30%) as unchanged drug and metabolites, and the remainder is cleared in feces.
3. Toxicity
a. Dose-limiting. Myelosuppression may be cumulative and recovery may be prolonged.
b. Occasional. Anorexia, nausea, vomiting, mucositis, sterility
c. Rare. Alopecia, pruritus, rash, hypersensitivity; secondary malignancies (acute leukemia); pulmonary fibrosis, vasculitis, cataracts
4. Administration
a. Supplied as 2-mg tablets
b. Dose modification. Hematologic; administer cautiously in patients with azotemia
c. Dose. If no myelosuppression is observed after oral dosing, poor oral absorption should be suspected. For continuous therapy: 0.10 to 0.15 mg/kg PO daily for 2 to 3 weeks, no therapy for 2 to 4 weeks, then 2 to 4 mg PO daily. For pulse therapy: 0.2 mg/kg (10 mg/m2) PO daily for 4 days every 4 to 6 weeks.
d. Drug interactions
(1) Cimetidine may result in reduced serum melphalan levels.
(2) Cyclosporine enhances the risk of renal toxicity from melphalan.
I. Nitrogen mustard (mechlorethamine, Mustargen)
1. Indication. Hodgkin lymphoma; topical use for T-cell lymphoma
2. Pharmacology
a. Mechanism. Rapid alkylation of DNA, RNA, and protein (see Section I.A). Cell cycle-nonspecific with activity in all phases of the cell cycle.
b. Metabolism. Native drug is highly active and is rapidly deactivated within the blood by rapid hydrolysis; the elimination half-life is 15 minutes. Metabolites are mostly excreted in urine.
3. Toxicity
a. Dose-limiting. Myelosuppression
b. Common. Severe nausea and vomiting beginning 1 hour after administration; skin necrosis if extravasated (sodium thiosulfate may be tried); burning at IV injection site and facial flushing; metallic taste; discoloration of the infused vein; abnormal LFTs within 1 week of therapy (up to 90% of patients)
c. Occasional. Alopecia, sterility, diarrhea, thrombophlebitis, gynecomastia
d. Rare. Neurotoxicity (including hearing loss), angioedema, secondary neoplasms
4. Administration. Patients should always be premedicated with antiemetics. The drug should be administered through the tubing of a running intravenous line using extravasation precautions.
a. Supplied as 10-mg vials
b. Dose modification. Hematologic; none required for hepatic or renal impairment
c. Dose. 10 mg/m2 as a single or divided dose monthly or 6 mg/m2 on day 1 and day 8 of the MOPP regimen (see Appendix D-1)
d. Drug interactions. Sodium thiosulfate inactivates mechlorethamine.
J. Nitrosoureas.
Carmustine [BCNU, bischlorethyl nitrosourea (BiCNU)]; lomustine [CCNU, cyclohexyl chlorethyl nitrosourea (CeeNU)]; streptozocin, which is a nitrosourea with a different mechanism of action (see Section I.K)
1. Indications. Brain cancer, myeloma, melanoma, and some carcinomas
2. Pharmacology
a. Mechanism. Alkylation of DNA and RNA (see Section I.A); DNA cross-linking; inhibition of DNA polymerase, DNA repair, and RNA synthesis. Cell cycle-nonspecific.
b. Metabolism. Highly lipid-soluble drugs that enter the brain. Rapid spontaneous decomposition to active and inert products; the drugs also are metabolized. Most of the intact drug and metabolic products are excreted in urine; some products have an enterohepatic cycle.
3. Toxicity
a. Dose-limiting. Myelosuppression is prolonged, cumulative, and substantially aggravated by concurrent radiation therapy.
b. Common. Nausea and vomiting may last 8 to 24 hours. BCNU causes local pain during injection or hypotension during a too rapid or concentrated injection.
c. Occasional. Stomatitis, esophagitis, diarrhea, LFT abnormalities; alopecia, facial flushing, brown discoloration of skin; interstitial lung disease with pulmonary fibrosis (with prolonged therapy and higher doses, especially with cumulative doses >1,400 mg/m2); dizziness, optic neuritis, ataxia, organic brain syndrome; renal insufficiency
d. Rare. Secondary malignancies
4. Administration. Avoid alcohol at least 1 hour before and after CCNU.
a. Supplied as 100-mg vials of BCNU; 10-, 40-, and 100-mg capsules of CCNU in a dose pack of 300 mg
b. Dose modification. Hematologic and renal
c. Dose
(1) BCNU: 150 to 200 mg/m2 IV (as asingle dose or divided over 2 days) every 6 to 8 weeks. Do not infuse over longer than 2 hours because of incompatibility of the drug with intravenous tubing. If blood and BCNU are mixed in the syringe before administration, the painfulness of injection may be decreased.
(2) CCNU: 100 to 130 mg/m2 PO every 6 to 8 weeks
d. Drug interactions
(1) With cimetidine to decrease nitrosourea metabolism, resulting in increased hematosuppression
(2) BCNU may lower levels of digoxin and phenytoin.
(3) Amphotericin B enhances cellular uptake of BCNU, resulting in increased host toxicity.
K. Procarbazine (N-methylhydrazine, Matulane, Natulan)
1. Indications. Hodgkin lymphoma, cutaneous T-cell lymphoma
2. Pharmacology
a. Mechanism. DNA alkylation and depolymerization. Methylation of nucleic acids. Inhibition of DNA, RNA, and protein synthesis.
b. Metabolism. Rapidly and extensively metabolized by the hepatic P450 microsomal system. Metabolic activation of the drug is required. Readily enters the cerebrospinal fluid. Degraded in the liver to inactive compounds, which are excreted in urine (70%). Less than 10% of the drug is excreted in unchanged form.
3. Toxicity
a. Dose-limiting. Myelosuppression, which is most pronounced 4 weeks after starting treatment
b. Common. Nausea and vomiting, which decrease with continued use; flu-like syndrome (usually with initial therapy); sensitizes tissues to radiation; amenorrhea and azoospermia, sterility
c. Occasional. Dermatitis, hyperpigmentation, photosensitivity; stomatitis, dysphagia, diarrhea; hypotension, tachycardia; urinary frequency, hematuria; gynecomastia
d. Neurologic. Procarbazine results in disorders of consciousness or mild peripheral neuropathies in about 10% of cases. These abnormalities are reversible and rarely serious enough to alter drug dosage. Manifestations of toxicity include sedation, depression, agitation, psychosis, decreased deeptendon reflexes, paresthesias, myalgias, and ataxia.
e. Rare. Xerostomia, retinal hemorrhage, photophobia, papilledema; hypersensitivity pneumonitis, secondary malignancy
4. Administration. Avoid alcohol, tyramine-containing foods, tricyclic antidepressants, antihistamines, dark beer, wine, cheeses, bananas, yogurt, and pickled or smoked foods.
a. Supplied as 50-mg capsules
b. Dose modification. Reduce dose in patients with hepatic, renal, or bone marrow dysfunction.
c. Dose. 60 to 100 mg/m2 PO daily for 10 to 14 days in combination regimens
d. Drug interactions. Procarbazine is a monoamine oxidase inhibitor and thus interacts with numerous agents. For the most part, these interacting agents should be avoided for about 2 weeks after stopping procarbazine. Potential reactions from procarbazine interactions with other drugs include the following:
(1) Disulfiram (Antabuse)-like reactions: Alcohol
(2) Severe hypertension
(a) Sympathomimetic amines, levodopa, methyldopa; cocaine, narcotics; buspirone, methylphenidate (Ritalin); dextromethorphan (with hyperpyrexia); caffeine
(b) Foods and beverages containing amines (e.g., aged cheese, beer, and wine [with or without alcohol]; smoked or pickled meats, poultry or fish; fermented sausage; any overripe fruit)
(3) Hypotension: Hypotension-producing medications, spinal anesthetics
(4) CNS depression and anticholinergic effects: Antihistamines, phenothiazines, barbiturates, and other CNS depressants
(5) Hyperpyrexia, convulsions, and death: Tricyclic antidepressants, monamine oxidase inhibitors, fluoxetine; sympathomimetic amines; meperidine and other narcotics (also possibly hypotension, respiratory depression, and coma)
(6) Hypoglycemia with insulin or sulfonylureas
(7) Increased anticoagulant effect with coumarin derivatives
(8) Shaking, hyperventilation, confusion, and so forth, with tryptophan
L. Streptozocin (streptozotocin, Zanosar)
1. Indications. Islet cell cancer of the pancreas (in combination with fluorouracil), carcinoid tumors
2. Pharmacology
a. Mechanism. Alkylating agent (see Section I.A). A cell cycle-nonspecific nitrosourea analog. Inhibits DNA synthesis and the DNA repair enzyme, guanine-O6-methyl transferase; affects pyrimidine nucleotide metabolism and inhibits enzymes involved in gluconeogenesis. Selectively targets pancreatic βcells, presumably due to the glucose moiety on the molecule.
b. Metabolism. Drug is a type of nitrosourea that is extensively metabolized by the liver to active metabolites and has a short plasma half-life (<1 hour). Crosses the blood-brain barrier. Excreted in urine as metabolites and unchanged drug.
3. Toxicity
a. Dose-limiting. Nephrotoxicity initially appears as proteinuria and progresses to glycosuria, aminoaciduria, proximal renal tubular acidosis, nephrogenic diabetes insipidus, and renal failure if the drug is continued.
b. Common. Nausea and vomiting (often severe), myelosuppression (mild, but may be cumulative), hypoglycemia after infusion, vein irritation during infusion, altered glucose metabolism with either hypoglycemia or hyperglycemia
c. Occasional. Diarrhea, abdominal cramps, LFT abnormalities
d. Rare. Central nervous system toxicity, fever, secondary malignancies
4. Administration. Urinalysis and serum creatinine levels are monitored before each dose. Patients are routinely premedicated with antiemetics. The dose is administered over 30 to 60 minutes to prevent local pain.
a. Supplied as 1-g vials
b. Dose modification. Proteinuria or elevated serum creatinine levels contraindicate use of the drug until the abnormalities resolve.
c. Dose. 1.0 g/m2 IV weekly for 6 weeks, then off treatment for 4 weeks, or 0.5 g/m2 IV daily for 5 days every 6 weeks
M. Temozolomide (methozolastone, Temodar, Temodal)
1. Indications. Brain tumors; metastatic melanoma
2. Pharmacology. Structurally and functionally similar to dacarbazine
a. Mechanisms. Structurally and functionally similar to dacarbazine. Metabolic activation to the reactive compound (MTIC) is required for antitumor activity. The drug methylates guanine residues in DNA and inhibits DNA, RNA, and protein synthesis, but does not cross-link DNA strands. Nonclassic alkylating agent, cell cycle-nonspecific.
b. Metabolism. Excreted predominantly by the renal tubules. Because the drug is lipophilic, it crosses the blood-brain barrier.
3. Toxicity
a. Dose-limiting. Myelosuppression
b. Common. Mild to moderately severe nausea and vomiting, diarrhea, headache, fatigue, mild transaminase elevation
c. Occasional. Photosensitivity, myalgias, fever
d. Rare. Prolonged cytopenia, myelodysplastic syndrome
4. Administration. Patients should avoid sun exposure during and for several days after treatment.
a. Supplied as 5-, 20-, 100-, 140-, 180-, and 250-mg capsules and 100 mg vials for IV injection
b. Dose modification. Consider dosage reduction for moderately severe hepatic or renal dysfunction and for elderly patients.
c. Dose. 75 mg/m2 PO daily during radiation therapy; 150 mg/m2 PO for 5 days each month as maintenance therapy, increasing to 200 mg/m2 if tolerated
N. Thiotepa (triethylenethiophosphoramide, Thioplex)
1. Indications. Intracavitary for malignant effusions, intravesicular for urinary bladder, and intrathecal use for meningeal metastasis; severe thrombocytosis. Also can be used for breast and ovarian cancers and for autologous hematopoietic stem cell transplantation.
2. Pharmacology. Ethylenimine analog, chemically related to nitrogen mustard
a. Mechanism. Alkylation (see Section I.A). Alkylates the N-7 position of guanine. Cell cycle-nonspecific.
b. Metabolism. Rapidly decomposed in plasma and excreted in urine. Extensively metabolized by the hepatic P450 microsomal system to active and inactive metabolites.
3. Toxicity
a. Dose-limiting. Myelosuppression, which may be cumulative
b. Common (for intravesicular administration). Chemical cystitis, abdominal pain, hematuria, dysuria, frequency, urgency, ureteral obstruction; nausea and vomiting 6 hours after treatment
c. Occasional. GI upset, abnormal LFTs, rash, hives; hypersensitivity
d. Rare. Alopecia, fever, angioedema, secondary malignancies
4. Administration. Thiotepa has been administered intravenously, intramuscularly, intravesicularly, intrathecally, intra-arterially, intrapleurally, intrapericardially, intraperitoneally, intratumorally, and as an ophthalmic instillation.
a. Supplied as 15-mg vials
b. Dose modification. Necessary for patients with cytopenias
c. Dose. 10 to 20 mg/m2 IV every 3 to 4 weeks; 30 to 60 mg intravesicularly every week for 4 weeks; 1 to 10 mg/m2 intrathecally twice weekly
d. Drug interactions. Increases neuromuscular blockade with succinyl choline
O. Cisplatin [cis-diamminedichloroplatinum (CDDP), Platinol]
1. Indications. A wide variety of malignancies
2. Pharmacology
a. Mechanism. A heavy metal alkylator of DNA. Covalently bonds to proteins, RNA, and especially DNA, forming DNA cross-linking and intrastrand N-7 adducts. The transisomer has virtually no antitumor activity. Acquired resistance to cisplatin involves alterations in transmembrane transport of drugs, intracellular levels of glutathione (GSH) or sulfhydryl-containing proteins, and the capacity to repair cisplatin DNA lesions.
b. Metabolism. Widely distributed in the body, except for the CNS. Long half-life in plasma (up to 3 days); may remain bound in tissues for months. Biliary excretion accounts for <10% of the total drug excretion. Approximately 15% of drug is excreted in the urine unchanged, and 10% to 40% of the remainder is excreted in the urine within 24 hours.
3. Toxicity
a. Dose-limiting
(1) Cumulative renal insufficiency. The incidence of renal insufficiency is about 5% with adequate hydration measures and 25% to 45% without hydration measures.
(2) Peripheral sensory neuropathy develops after the administration of 200 mg/m2 and can become dose-limiting when the cumulative cisplatin dose exceeds 400 mg/m2. Symptoms may progress after treatment is discontinued and include loss of proprioception and vibratory senses, hyporeflexia, and the Lhermitte sign. Symptoms may resolve slowly after many months.
(3) Ototoxicity with tinnitus and high-frequency hearing loss occurs in 5% of patients. Ototoxicity occurs more commonly in patients receiving doses of >100 mg/m2 by rapid infusion or high cumulative doses.
b. Common. Severe nausea and vomiting (both acute and delayed) occur in all treated patients; preventative antiemetic regimens are required. Hypokalemia, hypomagnesemia (occasionally difficult to correct), and mild myelosuppression occur very frequently; anorexia and metallic taste of foods; alopecia; azoospermia, sterility, impotence.
c. Occasional. Alopecia, loss of taste, vein irritation, transiently abnormal LFTs, SIADH, hypophosphatemia, myalgia, fever; optic neuritis
d. Rare. Altered color perception and reversible focal encephalopathy that often causes cortical blindness. Raynaud phenomenon, bradycardia, bundle-branch block, congestive heart failure; anaphylaxis, tetany.
4. Administration
a. Supplied as multidose vials
b. Dose modification. Renal function must return to normal before cisplatin can be given. Many physicians avoid using cisplatin when the creatinine clearance is <40 mL/min. Use with caution in patients with documented hearing impairment.
c. Dose depends on the chemotherapy regimen, of which there are many. Examples are
(1) 40 to 120 mg/m2 or more IV every 3 to 4 weeks, or
(2) 20 to 40 mg/m2 IV daily for 3 to 5 days every 3 to 4 weeks
d. Method. The principles of cisplatin administration are as follows:
(1) Monitoring. Serum creatinine, electrolytes, magnesium, and calcium levels should be measured daily during therapy. Audiometry is usually not necessary.
(2) Antiemetics. Patients should be given prophylactic antiemetics, such as ondansetron and dexamethasone.
(3) Hydration and diuresis are required when 40 mg/m2 or more of cisplatin is given to increase urine output before administration of the drug. Continue hydration (IV or PO) for 24 hours after the drug is given. Furosemide is given to prevent fluid overload. Intravenous fluids are supplemented with KCl and MgSO4.
(4) Amifostine cytoprotection (see Section X.A). Amifostine and mesna may inactivate the nephrotoxic effect of cisplatin.
e. Drug interactions
(1) Taxanes should be given before cisplatin when used in combination because cisplatin decreases taxane clearance when given immediately prior to taxanes.
(2) Nephrotoxicity risk may be increased with rituximab, aminoglycosides, and other nephrotoxic agents.
(3) Loop diuretics (e.g., bumetanide, furosemide) may potentiate ototoxicity.
(4) Neurotoxicity may be increased with altretamine or vinca alkaloids.
(5) Anticonvulsant (e.g., carbamazepine, phenytoin) serum levels may be decreased.
P. Carboplatin (Paraplatin)
1. Indications. A wide variety of malignancies
2. Pharmacology
a. Mechanisms. Heavy metal alkylating-like agent with mechanisms very similar to cisplatin, but with different toxicity profile. Like cisplatin, it produces predominantly interstrand DNA cross-links rather than DNA-protein cross-links; this effect is apparently cell cycle-nonspecific. Cisplatin and carboplatin exhibit substantial clinical cross-resistance.
b. Metabolism. Plasma half-life of only 2 to 3 hours. Excreted in urine as unchanged drug (70%) and metabolites.
3. Toxicity
a. Dose-limiting. Myelosuppression is significant and cumulative, especially thrombocytopenia. Median nadir hematosuppression at 21 days; increased myelosuppression in patients who have reduced creatinine clearance levels or who have received prior chemotherapy.
b. Common. Nausea, vomiting, myalgias, weakness, and nephrotoxicity (but less severe less common than with cisplatin); pain at injection site; cation electrolyte imbalance
c. Occasional. Reversible abnormal LFTs, azotemia; peripheral neuropathy (5%), visual disturbance; hypersensitivity reactions; amenorrhea, azoospermia, impotence, and sterility
d. Rare. Alopecia, rash, flu-like syndrome, hematuria, hyperamylasemia; hearing loss, optic neuritis; alopecia
4. Administration
a. Supplied as 50-, 150-, 450-mg vials
b. Dose modification. Reduce dosage for creatinine clearance of ≤60 mL/min. Caution is advised when concomitantly administering other myelosuppressive or nephrotoxic drugs.
c. Dose by creatinine clearance (ClearanceCr), as follows: ClearanceCr ≥60 mL/min; dose = 360 mg/m2 ClearanceCr ≥41 to 59 mL/min; dose = 250 mg/m2 ClearanceCr ≥16 to 40 mL/min; dose = 200 mg/m2
d. Dose by Calvert formula (AUC, area under the curve; GFR, glomerular filtration rate):
Total dose [mg (not per m2)] = (target AUC) × (GFR +25) Target AUC = 4 to 6 for previously treated patients Target AUC = 5 to 7 for previously untreated patients
e. Drug interactions.
(1) Taxanes should be generally administered before carboplatin when given concomitantly. However, unlike cisplatin, there is no significant interaction between paclitaxel and carboplatin.
(2) Anticonvulsant (e.g., carbamazepine, phenytoin) serum levels may be decreased.
(3) Aminoglycosides may increase nephrotoxicity.
Q. Oxaliplatin (diaminocyclohexane platinum, Eloxatin)
1. Indications. Colorectal, pancreatic, and gastric cancers
2. Pharmacology
a. Mechanisms. Binds covalently to DNA with preferential binding to the N-7 position of guanine and adenine; intrastrand and interstrand cross-links.
b. Metabolism. Undergoes extensive nonenzymatic conversion to its active cytotoxic species; >50% of the drug is cleared through the kidneys. Only 2% of the drug is excreted in feces.
3. Toxicity
a. Dose-limiting
(1) Acute dysesthesias in the hands, feet, perioral area, or throat develop within hours or up to 2 days after dosing, may be precipitated or exacerbated by exposure to cold (cold air or beverages); usually resolves within 2 weeks; frequently recurs with further dosing and may be ameliorated by prolonging the infusion to 6 hours. Dysphagia, dyspnea without stridor or wheezing, jaw spasms, dysarthria, voice changes, or chest pressure may occur. In contrast to cisplatin, ototoxicity occurs rarely.
(2) Persistent peripheral sensory neuropathy usually characterized by paresthesias, dysesthesias, and hypesthesia, including deficits in proprioception, which is usually reversible within 4 months of discontinuing oxaliplatin.
b. Common. Anorexia, nausea, vomiting, constipation, diarrhea, abdominal pain; fever, fatigue; mild to moderate myelosuppression; mild to moderate LFT abnormalities
c. Occasional. Allergic reactions, mild nephrotoxicity, headache, stomatitis, taste alteration; back pain, arthralgias
d. Rare. Pulmonary fibrosis
4. Administration. The drug cannot be mixed with alkaline medications or media [such as basic solutions of fluorouracil (5-FU)]. The patient should avoid exposure to cold.
a. Supplied as 50- and 100-mg vials
b. Dose modification. Reduce dose for renal dysfunction
c. Dose
(1) FOLFOX-4 regimen: 85 mg/m2 with leucovorin, 200 mg/m2, both given over 2 hours at the same time through Y-tubing on day 1. Then, 5-FU is given first as a bolus at a dose of 400 mg/m2 and then as an infusion of 600 mg/m2 over 22 hours. On day 2, leucovorin, 5-FU bolus, and 5-FU infusion over 22 hours are repeated. The cycle is repeated every 2 weeks. Several variations of this regimen are available.
(2) 100 to 130 mg/m2 IV every 3 weeks, either alone or in combination with other drugs.
II. ANTIMETABOLITES
A. General pharmacology of antimetabolites
1. Some antimetabolites are structural analogs of normal molecules that are essential for cell growth and replication. Other antimetabolites inhibit enzymes that are necessary for the synthesis of essential compounds. Their major effect is interfering with the building blocks of DNA synthesis (Fig. 4.1). Their activity, therefore, is greatest in the S phase of the cell cycle. In general, these agents have been most effective when cell proliferation is rapid.
2. The pharmacokinetics of these drugs are characterized by nonlinear doseresponse curves; after a certain dose, no more are killed with increasing doses (fluorouracil is an exception). Because of the entry of new cells into the cycle, the length of time that the cells are exposed to the drug is directly proportional to the killing potential.
B. Azacitidine (5-azacitidine, Vidaza)
1. Indication. Acute myelogenous leukemia (experimental); severe myelodysplastic syndromes (MDS) with an anticipated response in 16% of patients
2. Pharmacology
a. Mechanism. Antimetabolite (cytidine analog). Rapidly phosphorylated and incorporated into DNA and RNA, thereby inhibiting protein synthesis; also inhibits pyrimidine synthesis and DNA methylation.
b. Metabolism. Activated by phosphorylation and deactivated by deamination; similar to cytarabine. Excreted in urine (20% as unchanged drug).
3. Toxicity
a. Dose-limiting. Myelosuppression; nausea and vomiting.
b. Common. Hepatic dysfunction, fatigue, headache, diarrhea, alopecia, fever, injection site erythema
c. Occasional. Neurotoxicity (dizziness, restlessness, confusion), azotemia (transient), arthralgias, hypophosphatemia with myalgia, stomatitis, phlebitis, rash
d. Rare. Progressive lethargy and coma, renal tubular acidosis, rhabdomyolysis, hypotension
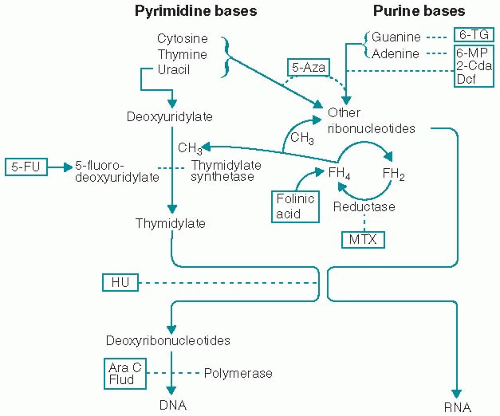 Figure 4.1. Sites of action of antimetabolites. 2-Cda, 2-chlorodeoxyadenosine; 5-Aza, 5-azacytidine; 5-FU, 5-fluorouracil; 6-MP, 6-mercaptopurine; 6-TG, 6-thioguanine; Ara C, cytosine arabinoside; Dcf, deoxycoformycin; Flud, fludarabine; HU, hydroxyurea; MTX, methotrexate; reductase, dihydrofolate reductase. |
4. Administration
a. Supplied as 100-mg vials
b. Dose modification. Necessary for patients with impaired liver function. Also reduce dose for patients with renal dysfunction and for serum bicarbonate concentration of <20 mEq/L.
c. Dose. 75 to 100 mg/m2 per day SQ for 7 days every 4 weeks for MDS (several cycles may be required for effectiveness)
d. Drug interactions. None known
C. Cladribine [2-chlordeoxyadenosine (2-CdA), Leustatin]
1. Indications. Hairy cell leukemia, indolent lymphoproliferadve disorders
2. Pharmacology. An analog of the purine deoxyadenosine
a. Mechanism. Antimetabolite. The analog accumulates in cells (particularly lymphocytes), blocks adenosine deaminase, and inhibits RNA and DNA synthesis. Inhibits ribonucleotide reductase. Depletes ATP. Induces apoptosis. Active against both dividing and resting cells.
b. Metabolism. Rapidly metabolized and eliminated through the kidneys
3. Toxicity. Patients are at increased risk for opportunistic infections.
a. Dose-limiting. Myelosuppression
b. Common. Immunosuppression with decreases in CD4+ and CD8+ cells; nausea, skin reactions at injection site; fever in 50% (most likely due to tumor’s releasing pyrogens and cytokines), chills, flu-like syndrome
c. Occasional. Neurotoxicity (headache, dizziness), hypersensitivity reactions, fatigue
d. Rare. Severe neurotoxicity, pancreatitis
4. Administration
a. Supplied as 10-mg vials
b. Dose modification. Hematologic; use with caution with renal dysfunction.
c. Dose. Either 0.10 mg/kg/d (4 mg/m2/d) by continuous IV infusion for 7 days, or 0.14 mg/kg daily IV over 2 hours for 5 days
d. Drug interactions. None known
D. Clofarabine (2-chloro-2/-fluorodeoxy-9-beta-D-arabinofurosyladenine, Clolar)
1. Indications. Relapsed or refractory acute lymphoblastic leukemia for patients between 1 and 21 years of age
2. Pharmacology. Purine antimetabolite
3. Toxicity
a. Dose-limiting.
(1) Capillary leak syndrome (CLS)/systemic inflammatory response syndrome (SIRS) is a development following cytokine release and manifested by hypotension, tachycardia, tachypnea, and pulmonary edema.
(2) Hematosuppression (90%)
(3) Hepatotoxicity and nephrotoxicity
b. Common. Tachycardia, hypotension, flushing; headache, fever, chills, fatigue; pruritus, rash; nausea, vomiting, diarrhea; abnormal LFTs (80%; generally occur within 10 days of administration with a duration of ≤2 weeks); increased creatinine (50%), limb pain
c. Occasional. Hypertension, edema, dyspnea, pleural, or pericardial effusion; mucositis; myalgia, arthralgia; irritability, somnolence, agitation; cecitis; CLS (4%), SIRS (2%)
d. Rare. Hepatic veno-occlusive disease, Stevens-Johnson syndrome, hallucination
4. Administration. Consider prophylactic corticosteroids, antiemetics, hydration, and allopurinol. Discontinue if hypotension develops during administration to ensure that it is not due to CLS/SIRS.
a. Supplied as 1 mg/mL in 20 mL vials
b. Dose modification: Use with caution in patients with renal or hepatic impairment. Withhold clofarabine and reinitiate with 25% dose reduction when returned to baseline for neutrophil counts <500/µL lasting ≥4 weeks, increase in creatinine or bilirubin (grade 3), or grade 3 GI toxicity. Discontinue clofarabine for CLS, SIRS, or grade 4 nonhematologic toxicity.
c. Dose for adults: 52 mg/m2 IV over 2 hours for 5 days every 2 to 6 weeks based on recovery from adverse effects or 40 mg/m2 IV over 2 hours for 5 days every 4 weeks
d. Drug interactions. No specific drug interactions have been reported. Avoid using potentially hepatotoxic or nephrotoxic drugs during the days of clofarabine administration.
E. Cytarabine (cytosine arabinoside, Cytosar, ara-C)
1. Indications. Acute leukemia, chronic myelogenous leukemia, lymphoma, meningeal involvement with tumor
2. Pharmacology. An analog of deoxycytidine
a. Mechanism. Antimetabolite. Requires intracellular activation to its phosphorylated derivative (ara-CTP), which inhibits DNA polymerases that are involved in the conversion of cytidine to deoxycytidine; some are incorporated into DNA. Ara-CTP inhibits ribonucleotide reductase, which results in decreased levels of deoxyribonucleotides for DNA
synthesis and function. Blocks DNA synthesis and repair and terminates DNA chain elongation. Cell cycle-specific (S phase). b. Metabolism. Requires activation to ara-CTP by kinase; deactivated by deaminase; ara-C is rapidly and completely deaminated in liver, plasma, and peripheral tissues; ara-C antitumor activity depends on relative amounts of kinase and deaminase in cells. In patients with renal insufficiency, one metabolite (uracil arabinoside) has the ability to produce high concentrations of ara-CTP, which may result in CNS toxicity. Excreted in urine as inactive metabolites.
synthesis and function. Blocks DNA synthesis and repair and terminates DNA chain elongation. Cell cycle-specific (S phase). b. Metabolism. Requires activation to ara-CTP by kinase; deactivated by deaminase; ara-C is rapidly and completely deaminated in liver, plasma, and peripheral tissues; ara-C antitumor activity depends on relative amounts of kinase and deaminase in cells. In patients with renal insufficiency, one metabolite (uracil arabinoside) has the ability to produce high concentrations of ara-CTP, which may result in CNS toxicity. Excreted in urine as inactive metabolites.
3. Toxicity
a. Dose-limiting. Myelosuppression
b. Common. Nausea, vomiting, mucositis, diarrhea (potentiated by the addition of an anthracycline); conjunctivitis (usually within the first 3 days of high-dose regimens, but reduced with prophylactic glucocorticoids eye drops); hydradenitis, arachnoiditis with intrathecal administration.
c. Neurotoxicity (cerebellar ataxia, lethargy, confusion) begins on the fourth or fifth day of infusion and usually resolves within 7 days. The incidence and severity of toxicity are related to the dose given (especially with total dose of >48 g/m2), the rate of infusion (least incidence for continuous infusions), age (particularly older than 60 years), sex (especially male), and the degree of hepatic or renal dysfunction (particularly with creatinine clearance of <60 mL/min). In some cases, it is irreversible or fatal.
d. Occasional. Alopecia, stomatitis, metallic taste, esophagitis, hepatic dysfunction (mild and reversible), pancreatitis, severe GI ulceration; thrombophlebitis; headache; rash, transient skin erythema without exfoliation.
Ara-C syndrome, described in pediatric patients, is an allergic reaction manifested by fever, flu-like syndrome, myalgias, bone pain, maculopapular rash, conjunctivitis, and occasional chest pain (corticosteroids are effective).
e. Rare. Sudden respiratory distress rapidly progressing to noncardiogenic pulmonary edema; pericarditis, cardiomegaly, tamponade; urinary retention.
4. Administration. Use prophylactic glucocorticoid eye drops for patients receiving high-dose regimens.
a. Supplied as 100-, 500-, 1,000-, and 2,000-mg vials
b. Dose modification. Use cautiously in patients with liver or renal disease or with risk factors for neurotoxicity.
c. Dose
(1) See Chapter 21 for use in patients with lymphoma and Chapter 25 for use in patients with acute leukemia.
(2) For intrathecal administration: 50 to 100 mg in 10-mL saline for 1 to 3 days weekly
(3) Low-dose regimen: 10 mg/m2 SC every 12 to 24 hours for 15 to 21 days
(4) High-dose therapy should be given over 1 to 2 hours
d. Drug interactions
(1) Ara-C antagonizes the efficacy of gentamycin and digoxin
(2) Increased risk of pancreatitis in patients previously treated with L-asparaginase
F. Decitabine (5-aza-2/-deoxycytadine, Dacogen)
1. Indications. Myelodysplastic syndromes, chronic myelomonocytic leukemia
2. Pharmacology. Decitabine is an analogue of the natural nucleoside 2′-decoxycytidine.
a. Mechanisms. Decitabine is believed to exert its antineoplastic effects by inhibition of DNA methyltransferase, causing hypomethylation of DNA and cellular differentiation or apoptosis.
b. Metabolism. The metabolic fate and route of elimination are not known.
3. Toxicity
a. Dose-limiting. Hematosuppression (nadir at 35 days, recovery at 35 to 50 days)
b. Common. Hematosuppression, fatigue, fever; nausea, constipation (35%), diarrhea; headache, arthralgias, rigors, edema, cough; hyperglycemia, hypokalemia, hypomagnesemia.
4. Administration. Premedicate with antinausea drugs.
a. Supplied as 50-mg vials as a lyophilized powder
b. Dose modification: Reduce dose to 11 mg/m2 if hematologic recovery requires more than 6 weeks. Decitabine should be discontinued if the serum creatinine is ≥2 mg/dL or SGPT or bilirubin is greater than or equal to twice the upper limit of normal. The dose should also be reduced for grade 3 or 4 nonmyelosuppressive toxicities.
c. Dose. 15 mg/m2 IV over 1 hour for 5 days. Cycles are repeated every 4 weeks. Weekly and various other dosing schedules are available. Responses may require three cycles of treatment.
d. Drug interactions. None known
G. Fludarabine (2-fluoroadenine arabinoside-5-phosphate, Fludara, Oforta)
1. Indications. Chronic lymphocytic leukemia, low-grade lymphomas, and cutaneous T-cell lymphomas
2. Pharmacology. The 5/-monophosphate analog of ara-A (arabinofuranosyladenosine). The 2-fluoro group on the adenosine ring renders this drug resistant to breakdown by adenosine deaminase (compare with cytarabine).
a. Mechanism. Antimetabolite with high specificity for lymphoid cells. Its active metabolite, 2-fluoro-ara-A, appears to act by inhibiting DNA chain extension, DNA polymerase-α, and ribonucleotide reductase. It has activity against both dividing and resting cells and induces apoptosis.
b. Metabolism. Metabolites and unchanged drug (25%) are excreted primarily in urine.
3. Toxicity
a. Dose-limiting. Myelosuppression, which may be cumulative; severe autoimmune hemolytic anemia that may or may not be responsive to corticosteroids
b. Common. Immunosuppression with decreases in CD4+ and CD8+ T cells in most patients and associated with increased risk for opportunistic infections (recovery may take more than a year); mild nausea and vomiting; fever with associated flu-like syndrome (25%); cough, weakness, arthralgia/myalgias
c. Occasional. Alopecia (mild), abnormal LFTs, tumor lysis syndrome
d. Rare.
(1) Stomatitis, diarrhea; dermatitis; chest pain, hypotension, interstitial pneumonitis; delayed neurotoxicity (usually high doses: somnolence, transient paresthesias, demyelination)
(2) Immune-mediated hematologic effects (autoimmune hemolytic anemia, immunologic thrombocytopenic purpura, acquired hemophilia, transfusion-associated graft-vs.-host disease)
4. Administration
a. Supplied as 50-mg vials (Fludara) and 10-mg tablets (Oforta)
b. Dose modification. Decrease dosage by 25% for patients with creatinine clearance of <10 to 49 mL/min and by 50% or more for clearance of <10 mL/min
c. Dose. 25 mg/m2 IV over 30 minutes or 40 mg/m2 PO daily for 5 consecutive days every 4 weeks; 25 to 30 mg/m2 IV for 3 days every 4 weeks when combined with other agents.
d. Drug interactions.
(1) Fludarabine may enhance cytotoxicity of cyclophosphamide, cisplatin, and mitoxantrone by inhibiting nucleotide repair mechanisms and of cytarabine by inducing expression of deoxycytidine kinase.
(2) The combination of fludarabine plus pentostatin has resulted in a high incidence of fatal pulmonary toxicity.
H. 5-Fluorouracil (5-FU, Adrucil)
1. Indications. Gastrointestinal, breast, pancreatic, and head and neck carcinomas
2. Pharmacology. A fluoropyrimidine analog
a. Mechanism. Antimetabolite. Requires activation to cytotoxic metabolite forms. Interferes with DNA synthesis by blocking thymidylate synthetase, an enzyme involved in the conversion of deoxyuridylic acid to thymidylic acid. Metabolites (e.g., FUTP) are incorporated into several RNA species, which thereby interfere with RNA function and protein synthesis. Incorporation of another metabolite (FdUTP) into DNA results in inhibition of DNA synthesis and function. It is cell-cycle S-phase-specific but acts in other cell cycle phases as well and is unique in having a log linear cell-killing action.
b. Metabolism. 5-FU rapidly enters all tissues, including spinal fluid and malignant effusions. The drug undergoes extensive intracellular activation by a series of phosphorylating enzymes and phosphoribosyl transferase, particularly dihydropyrimidine dehydrogenase. Most of the drug degradation occurs in the liver. Responsive tumors appear to lack degradation enzymes. Metabolism eliminates 90% of 5-FU. Inactive metabolites are excreted in urine, bile, and breath (as carbon dioxide). The elimination half-life is short, ranging from 10 to 20 minutes.
3. Toxicity is more common and more severe in patients with dihydropyrimidine dehydrogenase deficiency.
a. Dose-limiting. Myelosuppression (less common with continuous infusion); mucositis (more common with 5-day infusion); diarrhea.
b. Common. Nasal discharge; eye irritation, and excessive lacrimation due to dacryocystitis and lacrimal duct stenosis; dry skin, photosensitivity, and pigmentation of the infused vein.
c. Neurologic. Reversible cerebellar dysfunction, somnolence, confusion or seizures occurs in about 1% of patients. Symptoms usually disappear 1 to 6 weeks after the drug is discontinued, but they abate after the dose is reduced or even if the same dose is maintained.
d. Occasional. Esophagitis; hand-foot syndrome with protracted infusion (paresthesia, erythema, and swelling of the palms and soles); coronary vasospasm (particularly in patients with a prior history of myocardial ischemia); thrombophlebitis; nausea, vomiting.
e. Rare. Alopecia, dermatitis, loss of nails, dark bands on nails; blurred vision, “black hairy tongue” (hypertrophy of filiform papillae), anaphylaxis, fever.
4. Administration. 5-FU is given by IV bolus, IV infusion over 15 minutes, continuous IV infusion, arterial infusion, intracavitarily, topically, or orally. The use of ice chips in the mouth 15 minutes before and 15 minutes after IV bolus injections of 5-FU may reduce the incidence and severity of mucositis.
a. Supplied as 500- and 1,000-mg vials
b. Dose modification. Fluorouracil is withheld if the patient has stomatitis, diarrhea, evidence of infection, leukopenia, or thrombocytopenia; drug is resumed (perhaps at reduced dosage) when these problems have resolved.
(1) May be contraindicated in patients with active ischemic heart disease or a history of myocardial infarction within the previous 6 months
(2) Patients who experience unexpected grade 3 or 4 myelosuppression, gastrointestinal and/or neurologic toxicities with initiation of therapy may have an underlying deficiency in dihydropyrimidine dehydrogenase. Further testing to identify this pharmacogenetic syndrome should be considered under these circumstances. If enzyme deficiency is present, therapy with 5-FU must be discontinued immediately.
c. Dose. Fluorouracil is erratically absorbed orally. Several regimens have been used, including the following:
(1) 500 to 600 mg/m2 IV weekly for 6 weeks of every 8 weeks
(2) 425 to 450 mg/m2 IV daily for 5 days every 28 days
(3) 800 to 1,000 mg/m2/d by continuous infusion for 4 to 5 days every 28 days
(4) 200 to 400 mg/m2/d by continuous infusion indefinitely
d. Drug interactions
(1) Toxicity is enhanced by leucovorin (along with antitumor activity), methotrexate, trimetrexate, and phosphonacetyl-L-aspartic acid (PALA).
(2) Allopurinol inhibits activation of 5-FU and may result in decreased effectiveness.
(3) Thymidine and uridine decrease the host toxic effects of 5-FU.
I. Leucovorin (folinic acid, citrovorum factor, 5-formyl tetrahydrofolate)
1. Indications. Combined with 5-FU in treatment of colorectal and other adenocarcinomas; the rescue agent for antifol toxicity (e.g., methotrexate)
2. Pharmacology
a. Mechanism. Leucovorin is a tetrahydrofolic acid derivative that acts as a cofactor for carbon transfer reactions in the synthesis of purines and pyrimidines. It inhibits the effects of methotrexate and other dihydrofolate reductase antagonists. Leucovorin potentiates the cytotoxic effects of fluorinated pyrimidines (i.e., 5-FU and floxuridine) by increasing the binding of folate cofactor and activated 5-FU to thymidylate synthetase (TS) within the cells.
b. Metabolism. Metabolized intracellularly to the reduced folate, 5, 10-methylenetetrahydrofolate, which forms a ternary complex with the 5-FU metabolite FdUMP and TS. Excreted in urine as metabolites.
3. Toxicity. Potentiates the toxic effects of fluoropyrimidine therapy
4. Administration
a. Supplied as 50-, 100-, 200-, 350-, and 500-mg vials for IV or IM use and as 5- and 15-mg tablets for oral use
b. Dose. Depends on combination regimen
(1) When used as a rescue agent in combination with high-dose methotrexate, leucovorin should be administered 24 hours after methotrexate every 6 hours for up to 12 doses, depending on the serum methotrexate level; continue leucovorin until the methotrexate level falls below 5 × 10-8 M.
(2) When given in combination with 5-FU, leucovorin should be administered at least 30 to 60 minutes before 5-FU to allow sufficient time for intracellular metabolism to take place.
c. Drug interactions. Barbiturates and phenytoin may decrease in their efficacy and increase the risk of seizures. Precipitates when mixed in the same solution as 5-FU.
J. Capecitabine (Xeloda)
1. Indications. Carcinomas of the breast or colon
2. Pharmacology. Capecitabine is a fluoropyrimidine carbamate that is a systemic prodrug of 5′-deoxy-5-fluorouridine (5′-DFUR), which is converted in vivo to 5-FU.
a. Mechanism. See fluorouracil
b. Metabolism. Hepatic. Catabolism predominantly via dihydropyrimidine dehydrogenase, which is present in liver, leukocytes, kidney, and other extrahepatic tissues. More than 90% is cleared in the urine (see 5-fluorouracil).
3. Toxicity. Similar to 5-FU
a. Dose-limiting. Diarrhea (50%), hand-foot syndrome
b. Common. Hand-foot syndrome (palmar-plantar erythrodysesthesia or chemotherapy-induced acral erythema) occurs in 15% to 50% of patients; nausea, vomiting, hematosuppression; fatigue.
c. Occasional. Abnormal LFTs, neurotoxicity; cardiac ischemia in patients with a prior history of coronary artery disease; tear duct stenosis, conjunctivitis, blepharitis; confusion, cerebellar ataxia.
4. Administration. Pyridoxine, 50 mg PO b.i.d. may be used to reduce the incidence and severity of the hand-foot syndrome. Celecoxib (Celebrex), 200-mg b.i.d., or a low-dose nicotine patch may also be effective.
a. Supplied as 150- and 500-mg tablets
b. Dose modification. Use with caution with liver dysfunction and in patients taking coumarin derivatives. Reduce dosage in patients with moderate renal dysfunction. Contraindicated in patients with dihydropyrimidine dehydrogenase deficiency or with severe renal impairment.
c. Dose. 650 to 1,250 mg/m2 PO b.i.d. (approximately every 12 hours) with a glass of water and within 30 minutes of a meal for 14 days every 3 weeks
d. Drug interactions
(1) Patients using warfarin should have dosage monitored closely, even after capecitabine is discontinued.
(2) Phenytoin toxicity can develop; dosage adjustment may be necessary.
(3) Liquid antacids may increase the bioavailability of capecitabine.
(4) Leucovorin enhances the antitumor effect and toxicity of capecitabine.
e. Treatment of hand-foot syndrome. Hand moisturizers; soak hands and feet in cool to tepid water for 10 minutes, then apply petrolatum jelly onto the wet skin. Bag balm or lanolin-containing salves may help.
K. Gemcitabine (Gemzar)
1. Indications. Carcinoma of pancreas, bladder, lung, ovary; soft tissue sarcomas.
2. Pharmacology. A fluorine-substituted deoxycytidine analog
a. Mechanisms. Cell-phase specific, primarily killing cells in S phase and also blocking the progression of cells through the G1 phase to S-phase boundary. Metabolized intracellularly to the active diphosphate and triphosphate. Inhibits ribonucleotide reductase; competes with deoxycytidine triphosphate (dCTP) for incorporation into DNA.
b. Metabolism. Undergoes extensive metabolism by deamination in the liver, plasma, and peripheral tissues. Nearly entirely excreted in urine as active drug and metabolites.
3. Toxicity
a. Dose-limiting. Myelosuppression
b. Common. Nausea, vomiting, diarrhea, stomatitis; fever with flu-like symptoms (40%); macular or maculopapular rash; transient LFT elevations; mild proteinuria and hematuria.
c. Occasional. Hair loss, rash, edema.
d. Rare. Hemolytic-uremic syndrome; pulmonary drug toxicity; hypersensitivity reactions; alopecia.
4. Administration. Gemcitabine is a potent radiosensitizer and should be avoided in patients while undergoing radiotherapy.
a. Supplied as vials of 200 and 1,000 mg
b. Dose modification. Use with caution in patients with hepatic or renal insufficiency.
c. Dose. 1,000 mg/m2 over 30 minutes weekly for 7 weeks or until toxicity, followed by 1 week rest; then for 3 of every 4 weeks.
d. Drug interactions. None known
L. Hydroxyurea (hydroxycarbamide, Hydrea, Droxia)
1. Indications. Myeloproliferative disorders, refractory ovarian cancer, sickle cell disease
2. Pharmacology. An analog of urea
a. Mechanism. Antimetabolite. Inhibits DNA synthesis by inhibiting nucleotide reductase, the enzyme that converts ribonucleosides to deoxyribonucleosides. Inhibits DNA repair and thymidine incorporation into DNA. Cell-cycle S-phase-specific, but acts in other phases as well.
b. Metabolism. Crosses the blood-brain barrier. Half of the drug is rapidly degraded into inactive compounds by the liver. Inactive products and unchanged drug (50%) are excreted in urine.
3. Toxicity
a. Dose-limiting. Myelosuppression, which recovers rapidly when treatment is stopped (prominent megaloblastosis)
b. Occasional. Nausea, vomiting, diarrhea; skin rash, facial erythema, hyperpigmentation; azotemia, proteinuria; transient LFT abnormalities; radiation recall phenomenon.
c. Rare. Alopecia, mucositis, diarrhea, constipation; neurologic events; pulmonary edema; flu-like syndrome; painful perimalleolar ulcers; possible acute leukemia in myeloproliferative disorders.
4. Administration
a. Supplied as 500-mg capsules (Hydrea); 200-, 300-, and 400-mg capsules (Droxia)
b. Dose modification. The drug should be given cautiously in the presence of liver dysfunction or when combined with other antimetabolites. Dosages should be reduced for creatinine clearance levels of <50 mL/min and when given with concomitant radiotherapy.
c. Dose. 10 to 30 mg/kg PO daily
d. Drug interactions
(1) Antiretroviral agents: hepatotoxicity and severe neurotoxicity
(2) Didanosine: pancreatitis
(3) 5-Fluorouracil: neurotoxicity
M. 6-Mercaptopurine (6-MP, Purinethol)
1. Indication. Acute lymphoblastic leukemia (maintenance therapy)
2. Pharmacology
a. Mechanism. Purine analog with activity in the S phase of the cell cycle. Inhibits de novo purine synthesis by inhibiting 5-phosphoribosyl-1-pyrophosphate. The parent drug is inactive. Requires intracellular phosphorylation by hypoxanthine-guanine phosphoribosyltransferase (HGPRT) to the monophosphate form, which is eventually metabolized to the triphosphate metabolite. Competes with ribotides for enzymes responsible for conversion of inosinic acid to adenine and xanthine ribotides. Its incorporation into DNA or RNA is of uncertain significance.
b. Metabolism. Mercaptopurine is slowly degraded in the liver, largely by xanthine oxidase. Allopurinol, a xanthine oxidase inhibitor, causes marked increase in its toxicity. Clearance is primarily hepatic with conventional doses.
3. Toxicity
a. Dose-limiting. Myelosuppression
b. Common. Mild nausea, vomiting, anorexia (25%); usually reversible cholestasis (30%); dry skin, photosensitivity; immunosuppression.
c. Rare. Stomatitis, diarrhea, dermatitis, fever, hematuria, Budd-Chiarilike syndrome, hepatic necrosis
4. Administration
a. Supplied as 50-mg tablets
b. Dose modification. Dose is reduced by 50% to 75% for patients with hepatic dysfunction.
c. Dose. 70 to 100 mg/m2 PO daily until patient responds or toxic effects are seen; then adjust for maintenance therapy.
d. Drug interactions. If given allopurinol, the 6-MP dose must be reduced by 75%. Dosage may also need to be modified if other hepatotoxic drugs are given. Warfarin dosages may be affected by 6-MP. Bactrim-DS may enhance myelosuppressive effect of 6-MP.
N. Methotrexate (amethopterin, MTX)
1. Indications. A wide variety of conditions
2. Pharmacology
a. Mechanism. Cell cycle-specific antifolate analog active in the S phase of the cell cycle. MTX blocks the enzyme dihydrofolate reductase, preventing formation of reduced (tetrahydro-) folic acid; tetrahydrofolic acid is crucial to the transfer of carbon units in a variety of biochemical reactions (Fig. 4.1). MTX thus blocks formation of thymidylate from deoxyuridylate and prevents synthesis of DNA. The drug also inhibits RNA and protein synthesis and prevents cells from entering the S phase of the cell cycle.
b. Metabolism. MTX is minimally metabolized by the human species. It is converted in the liver and other cells to higher polyglutamate forms. The drug is distributed to body water; patients with significant effusions eliminate the drug much more slowly. Because 50% to 70% of the drug is bound to plasma proteins, displacement by other drugs (e.g., aspirin, sulfonamides) may result in an increase in toxic effects. About 20% of the drug is eliminated in the bile. It is excreted in urine as unchanged drug (80% to 90% within 24 hours). Renal dysfunction results in dangerous blood levels of MTX and possible further renal damage. The half-life of the drug is 8 to 10 hours.
3. Toxicity. Leucovorin can reverse the immediate cytotoxic effects of MTX; generally, 1 mg of leucovorin is given for each 1 mg of MTX.
a. Dose-limiting. Myelosuppression, stomatitis, renal dysfunction
b. High-dose regimens. Nausea, vomiting, renal tubular necrosis, cortical blindness
c. Previously irradiated areas. Skin erythema, pulmonary fibrosis, transverse myelitis, cerebritis
d. Chronic therapy. Liver cirrhosis (reversible hepatic dysfunction occurs with short-term intermittent therapy); osteoporosis (in children).
e. Neurotoxicity. MTX neurotoxicity depends on dose and route of administration. Within a few hours after intrathecal administration, MTX can produce acute aseptic meningitis that is usually self-limited. A subacute encephalopathy and myelopathy can also occur after intrathecal administration.
High-dose systemic administration can cause a reversible encephalopathy of rapid onset and resolution that lasts from minutes to hours (stroke-like episodes). Chronic intrathecal combined with high-dose systemic administration can produce a more serious and irreversible leukoencephalopathy that develops months after treatment, is more likely to occur after brain irradiation, and causes dementia, seizures, spasticity, and ataxia.
f. Occasional. Nausea, vomiting, diarrhea (GI ulceration, hemorrhage, and perforation can occur if therapy is continued after the onset of diarrhea); dermatitis, photosensitivity, altered pigmentation, furunculosis; conjunctivitis, photophobia, excessive lacrimation, cataracts; fever, reversible oligospermia, flank pain (with rapid intravenous infusion).
g. Rare. Alopecia, MTX pneumonitis (see Chapter 29, Section IV.A)
4. Administration
a. Supplied as 2.5-, 5-, 7.5-, 10-, and 15 mg tablets and 20-to 1,000-mg vials
b. Dose modification. The drug must not be administered to any patient with a creatinine clearance level of <60 mL/min (serum creatinine >1.5 mg/dL).
c. Dose. Varies according to regimen
(1) High-dose regimens use supralethal doses of MTX followed by administration of the antidote leucovorin. This treatment is complex and requires experience for the clinician and use of special monitoring techniques.
(2) Intrathecal administration: 5 to 10 mg/m2 (maximum, 15 mg) in 7 to 15 mL of preservative-free saline (3 mL if given using an Ommaya reservoir) every 3 to 7 days
d. Drug interactions
(1) Leucovorin rescues normal tissues from MTX toxicity and may impair its antitumor activity. Folic acid supplements should be discontinued while on therapy.
(2) L-Asparaginase and thymidine also block MTX toxicity and antitumor action.
(3) Aspirin, other nonsteroidal anti-inflammatory agents, penicillins, cephalosporins, phenytoin, and probenecid decrease renal clearance of MTX and increase its toxicity.
(4) Sulfonamides and phenytoin displace MTX from protein-binding sites and may enhance its toxicity.
(5) Trimethoprim is also an inhibitor of dihydrofolate reductase and can enhance MTX toxicity.
(6) Parenteral acyclovir and concomitant intrathecal MTX may result in neurologic abnormalities.
(7) MTX may increase serum levels of warfarin, which is displaced from plasma proteins.
(8) Omeprazole (Prilosec) increases serum MTX levels.
O. Nelarabine (Arranon)
1. Indications. Relapsed or refractory T-cell acute lymphoblastic leukemia (ALL) and T-cell lymphoblastic lymphoma
2. Pharmacology
a. Mechanisms. A prodrug of ara-G, nelarabine is demethylated by adenosine deaminase to ara-G and then converted to ara-GTP, which is incorporated into the DNA of the leukemic blasts, leading to inhibition of DNA synthesis and inducing apoptosis. Ara-GTP accumulates at higher levels in T-cells, which correlates to clinical response.
b. Metabolism. Demethylated and hydrolyzed; drug and metabolites excreted in the urine.
3. Toxicity
a. Dose-limiting. Severe neurotoxicity that may not return to baseline after treatment cessation (discontinue drug for grade ≥ 2)
b. Common. Neurologic (70%; somnolence, confusion, dizziness, ataxia, tremor, peripheral neuropathy; severe neurotoxicity is reported including coma, demyelination, seizures, etc.); hematosuppression; fever, fatigue; nausea, vomiting, diarrhea, constipation; cough, edema
c. Occasional. Myalgia/arthralgia, abdominal pain, limb pain; stomatitis, dyspnea, cough; elevated transaminases or creatinine, hyper-/hypoglycemia
4. Administration.
a. Supplied as 5 mg/mL (50 mL vials)
b. Dose modification: Use with caution with hepatic or renal dysfunction; consider treatment delay for nonneurologic toxicity.
c. Dose (adults): 1,500 mg/m2/dose IV over 2 hours on days 1, 3, and 5; repeat every 21 days.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


