Tumor type
Approximate frequency (%)
Clinical characteristics
Imaging characteristics
Predominant pathology
Diffuse intrinsic
75–85
Multiple bilateral CN deficits
LTS
Ataxia
Short clinical history
Diffuse pontine enlargement
T1 hypointensity
T2 variable intensity
Little contrast enhancement
Astrocytoma (grades II–IV)
Focal midbrain
5–10
Signs and symptoms of raised ICP
Isolated CN deficit
Ataxia
Hemiparesis (rarer)
Torticollis
Small, well circumscribed
No edema
T1 hypointensity
T2 hyperintensity
Variable enhancement
Ventriculomegaly
Low-grade astrocytoma (grades I and II)
Ganglioglioma
Dorsally exophytic
10–20
Signs and symptoms of raised ICP
CN dysfunction
Prominent nystagmus
Torticollis
FTT (infants)
LTS typically absent
Arise from floor of fourth ventricle
T1 hypointensity
T2 hyperintensity
Bright enhancement
Pilocytic astrocytoma (grade I)
Grade II astrocytoma
Cervicomedullary
5–10
Lower CN dysfunction
LTS
Apnea
Sensory loss
Torticollis
Hydrocephalus (rarer)
Arise from lower medulla/upper cervical cord
Bulges dorsally toward fourth ventricle
T1 hypointensity
T2 hypointensity
Commonly enhances
Low-grade astrocytoma (WHO grade I or II)
Ganglioglioma
The differential diagnosis of brainstem gliomas can include vascular malformations, multiple sclerosis, and brainstem encephalitis, although high-quality MR imaging will frequently distinguish these. Rare tumors that can arise in the brainstem include primitive neuroectodermal tumor (PNET), atypical teratoid-rhabdoid tumor, lymphoma, ganglioglioma, and oligodendroglioma. Hemangioblastomas can present in association with von Hippel-Lindau disease (Jallo et al. 2004; Donaldson et al. 2008).
3.2 Focal Brainstem Gliomas
3.2.1 Epidemiology
Focal brainstem gliomas account for 20–35 % of childhood brainstem gliomas (Ramos et al. 2013; Guillamo et al. 2001). Because of their pathology and clinical features, they tend to have a longer latency period prior to clinical presentation; on average, the symptom duration prior to diagnosis is more than a year. In addition, the age at presentation tends to be older than diffuse brainstem gliomas with a mean age of 9 years and 11 months in one case review (Farmer et al. 2001). Focal brainstem gliomas occur more frequently in patients with neurofibromatosis type 1 (NF1) although their clinical features may be more benign than similar appearing lesions in patients without NF1 (Ramos et al. 2013; Ullrich et al. 2007; Pollack et al. 1996). The overall survival of these lower-grade gliomas is close to 80 % although substantial morbidity can occur depending upon their exact location within the brainstem and whether surgical resection is possible.
3.2.2 Pathology
The majority of the focal brainstem gliomas occur outside of the pons and are almost always low-grade astrocytomas. As with other glial tumors, the degree of cellularity and presence of nuclear atypia is used to further refine the histologic grade. Rosenthal fibers and microcystic structures support the diagnosis of pilocytic astrocytoma (WHO grade I), whereas WHO grade II tumors can lack these histologic signs and usually demonstrate higher degree of infiltrative behavior (Cillekens et al. 2000). Paradoxically, pilocytic astrocytomas can harbor features, such as mitotic activity and vascular proliferation, that would typically be associated with higher-grade glial tumors. Careful neuropathologic evaluation and correlation with imaging features are required to reach the correct diagnosis.
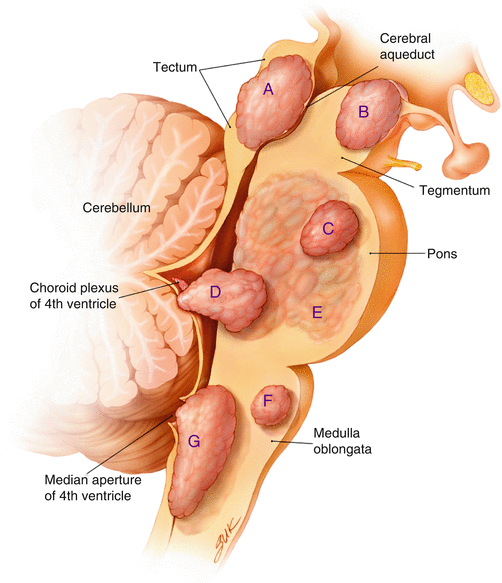
Focal brainstem gliomas are classified into three subtypes depending on their location: focal midbrain, dorsally exophytic from the pons, and cervicomedullary (Fig. 3.1). Midbrain and cervicomedullary tumors tend to be WHO grade I, whereas dorsally exophytic tumors tend to be WHO grade II. The reason for this variability depending on location in the brainstem is not known.
Similar to other lower-grade glial tumors such as fibrillary astrocytomas, gangliogliomas, and pleomorphic xanthroastrocytomas, low-grade brainstem gliomas can also harbor the BRAF V600E mutation (Schindler et al. 2011). They can also frequently harbor BRAF–KIAA1549 gene fusions, a hallmark feature of pilocytic astrocytomas (Jones and Baker 2014).
Isocitrate dehydrogenase 1 (IDH1) mutations have been identified in adolescents and young adults who present with brainstem astrocytomas. Patients with this mutation have tumors that exhibit less-aggressive biologic behavior, and those patients have a prolonged survival (Qi et al. 2014; Roberson et al. 2011).
3.2.3 Clinical Features
The clinical presentation of focal brainstem tumors can often be attributed to the site of origin (Table 3.1 and Fig. 3.1). Focal midbrain tumors tend to remain within the tectum of the midbrain, although they distort and compress the surrounding structures such as the cerebral aqueduct. Obstruction of the aqueduct of Sylvius will cause hydrocephalus leading to a common group of symptoms including headache, vomiting, visual disturbance, and gait instability. Figure 3.2 Dorsally exophytic tumors can either grow into the fourth ventricle causing obstructive hydrocephalus or, if they extend in the anterolateral direction into the cerebellopontine angle, cranial neuropathies. If the tumor is bulky and causes mass effect upon the cerebellum, symptoms of ataxia, nystagmus, and dysmetria can occur. Cervicomedullary tumors originate from the medulla and also tend to grow in a dorsal direction (Fig. 3.3). Their location leads to lower cranial nerve neuropathies.
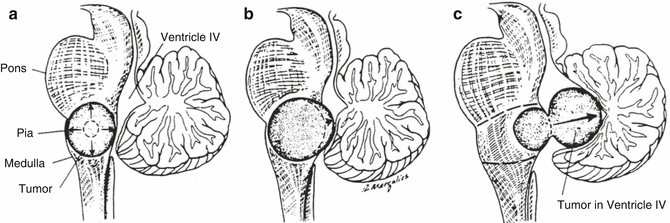
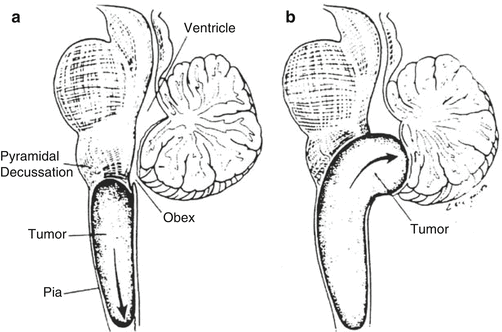
Fig. 3.2
(a–c) Illustrations of growth patterns of benign medullary tumors. (a) Focal medullary tumor displaces axially oriented fibers as it grows (arrows). (b) Larger focal medullary tumor tends to grow subependymally (arrowhead) because barriers limit its axial growth. (c) Subependymal lesion becomes dorsally exophytic (arrow) because of the limited resistance to growth offered by the ependyma (Reprinted with permission from Epstein and Farmer 1993)
Fig. 3.3
(a, b) Illustrations of growth patterns of cervicomedullary lesions. (a) Caudal growth is cylindrical, as for spinal cord tumors (arrow). (b) Rostral growth is directed toward the obex (arrow) as a result of hindrance from pial elements and decussating fibers (Reprinted with permission from Epstein and Farmer 1993)
3.2.4 Imaging
The radiologic features of focal brainstem gliomas can help differentiate it from diffuse gliomas and help further classify the extent of extension of the tumor beyond what the clinical signs may demonstrate. The tumors tend to be smaller and can contain cystic components. They are well demarcated without signs of infiltration and without associated edema (Ramos et al. 2013).
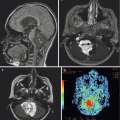
On computed tomography, focal midbrain tumors are isodense with the gray matter, whereas magnetic resonance imaging (MRI) typically shows an isointense or hypointense tumor on T1-weighted sequences and hyperintense on T2-weighted sequences (Fig. 3.4). Although focal midbrain tumors can extend beyond the tectum toward the thalamus or rostrally toward the pons, the edges of the tumor usually remain well defined. These tumors can contain calcifications and enhance poorly. They are uniform in appearance and typically do not enhance following administration with gadolinium.
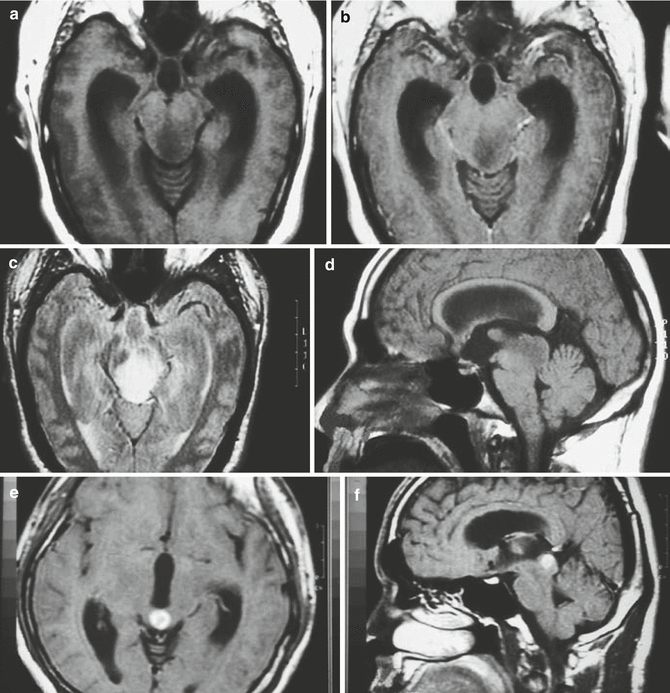
Fig. 3.4
(a–f) Magnetic resonance (MR) images of tectal gliomas. (a) Axial T1-weighted image without contrast: the lesion is small, is well circumscribed, and produces no edema. (b) Axial T1-weighted image following contrast: note the lack of enhancement. (c) Axial proton density image: the lesion is notably hyperintense, which contrasts with the hypointense signal of the lesion on T1-weighted pre-contrast images. (d) Sagittal T1-weighted image without contrast: the lesion is hypointense and does not produce significant edema. (e) Axial T1-weighted image following contrast: this tectal glioma exhibits obvious contrast enhancement, illustrating the enhancement variability of tectal gliomas. (f) Sagittal T1-weighted image following contrast: the lesion is well circumscribed and enhances brightly, which contrasts with the lesion noted in image (b)
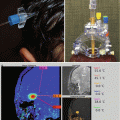
Dorsally exophytic tumors tend to grow only toward the ventricle rather than infiltrating or extending further into the brainstem (Fig. 3.5). The overwhelming majority of these tumors show either focal growth or exophytic growth (Farmer et al. 2001). In one case study, 50 % of the focal brainstem gliomas had uniform enhancement, which typically is not observed in diffuse pontine tumors where patchy enhancement is observed (Farmer et al. 2001). Future contrast enhancement may suggest local progression or growth during the course of monitoring for these tumors. Magnetic resonance spectroscopy has been evaluated for prognostic value. Of those tumors with a higher choline to N-acetylaspartate (Cho:NAA) ratio (greater than 1), there was a higher proportion that showed progression (Lazareff et al. 1998).
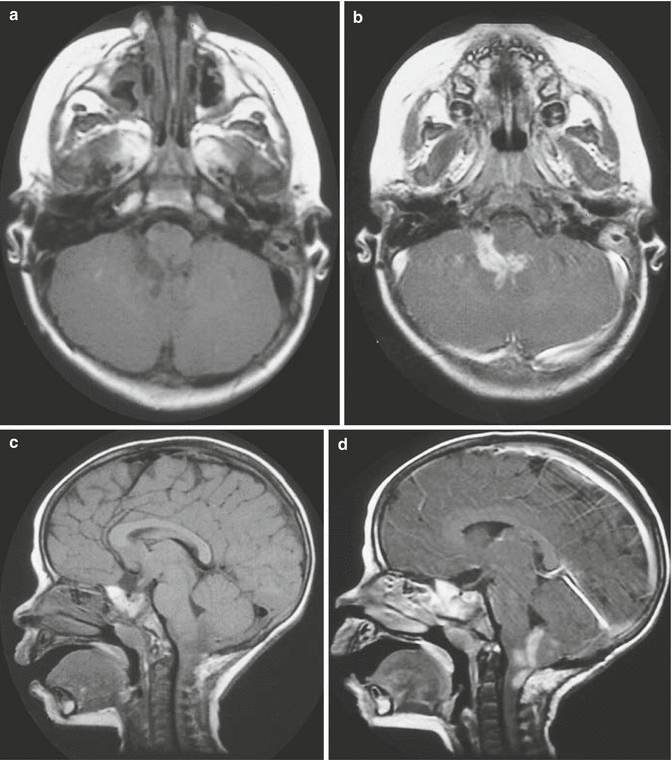
Fig. 3.5
(a–d) Magnetic resonance (MR) images of dorsally exophytic tumors. (a) Axial T1-weighted image without contrast: the lesion arises from the medulla and is hypointense on T1-weighted images. (b) Axial T1-weighted image with contrast: the lesion enhances brightly with contrast, which is a typical characteristic for dorsally exophytic tumors. (c) Sagittal T1-weighted image without contrast: the lesion does not invade the intrinsic tissue of the lower brainstem. (d) Sagittal T1-weighted with contrast: the enhancing lesion extrinsically involves the posterior upper cervical cord, medulla, and fourth ventricle
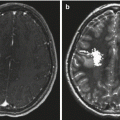
Cervicomedullary tumors appear as solid masses within the lower medulla and upper cervical cord that frequently extend into the fourth ventricle. Like the other brainstem tumors, they are hypointense on T1-weighted and hyperintense on T2-weighted images. They often enhance homogeneously with gadolinium (Fig. 3.6).
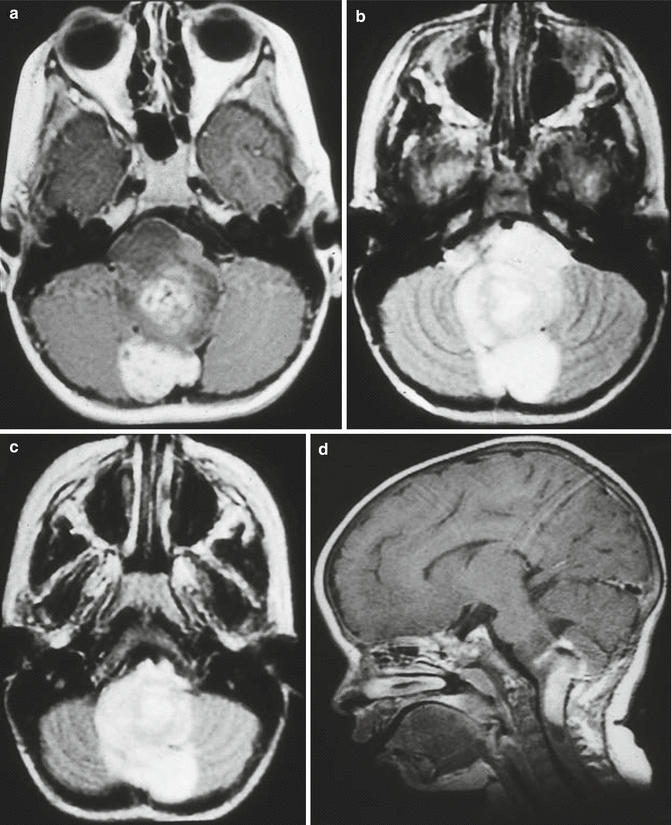
Fig. 3.6
Magnetic resonance (MR) images of cervicomedullary tumors. (a) Axial T1-weighted image with contrast: the lesion enhances heterogeneously and arises from the upper cervical cord. (b, c) Axial proton density images: the lesion is hyperintense. (d) Sagittal T1-weighted image following contrast: the lesion bulges dorsally into the fourth ventricle
3.2.5 Treatment
As these low-grade tumors progress slowly over time, symptomatic management is the mainstay of therapy, although surgical resection is indicated in specific circumstances. Focal midbrain tumors of the tectum can usually be identified with confidence by the MR imaging features. Initial symptom management includes treatment of hydrocephalus by endoscopic third ventriculostomy (preferred) or placement of a VP shunt. Since these tumors have an intrinsic location within the tectum, complete resection is usually not feasible without resulting in substantial morbidity. Following treatment of the hydrocephalus, observation with regular imaging studies is the primary management plan, and the tumor often remains stable for years. If there is late progression, a surgical biopsy may be needed to confirm the diagnosis prior to initiation of therapy. Of note, as cystic structures can often produce a more rapid onset of symptoms and cause morbidity, cyst removal can be a therapeutic option for these otherwise indolent tumors.
Dorsally exophytic tumors have a variable relationship to the adjacent brainstem. In some cases, the tumor-brain interface is well defined and the tumor can be resected with acceptable or minimal morbidity. Other tumors in this category have a poorly defined boundary and only subtotal resection can be performed. If there is an accessible location, an attempt at surgical resection is usually indicated. Subtotal resection will usually prompt further adjuvant. Cervicomedullary tumors are typically pilocytic astrocytomas (astrocytoma, WHO grade I) and as such often have a better plane between the tumor and the neural tissue. If the tumor reaches or is close to the dorsal pial surface, then resection should be considered. The surgical morbidity should be carefully considered prior to any surgery in view of the potential complications that would result in permanent injury to structures in this location.
Radiation therapy with 54 Gy in once daily fractions can control these focal lesions but carry a significant morbidity in pediatric patients despite the fact that focal glioma patients are older than those with diffuse gliomas. Chemotherapy options have been investigated including nitrosourea-based regimens, vinblastine-containing regimens, and carboplatin-containing regimens all with a high proportion showing disease stabilization (Gururangan et al. 2002; Bouffet et al. 2012; Jackacki et al. 2011). Temozolomide, which has been used in adult gliomas, has been used in this patient population showing tolerability and most patients having stable disease (Nicholson et al. 2007; Khaw et al. 2007). More recently, antiangiogenic therapy with irinotecan and bevacizumab produced disease control in those with recurrence of their focal brainstem tumors (Gururangan et al. 2014). Chemotherapeutic options can be used as a bridge to delay radiation.
3.3 Diffuse Brainstem Gliomas
3.3.1 Epidemiology
Virtually all gliomas in the diffuse category occur as expansile mass lesions within the ventral pons and are known as diffuse intrinsic pontine gliomas (DIPG). The incidence of DIPG in children in the United States is 300 cases a year in children, with an additional 100 arising in adults. The median age of presentation is between 5 and 9 years (Walker et al. 2004), and the incidence is equal among males and females (Jallo et al. 2004). They account for 80 % of all brainstem gliomas (Guillamo et al. 2001).
The average overall survival is 9–12 months from the time of diagnosis with the median time to death approximately 10 months (Kaplan et al. 1996). At 2 years, the overall survival is 20 %. A coexisting diagnosis of NF1 leads to an improved prognosis in tumors meeting similar diagnostic criteria – just as focal brainstem tumors carry an improved prognosis in these patients. Lack of cranial nerve involvement and a long latency period between the onset of symptoms and diagnosis carry an improved prognosis although retrospective data may suggest that the diagnosis in these patients is not that of DIPG (Kaplan et al. 1996). Lastly, children who are diagnosed with DIPG at less than 3 years of age can have a prolonged survival. These younger patients receiving similar therapy (including radiation therapy) have a 3-year progression free survival of 45 ± 19 % (Broniscer et al. 2008). There have been case reports of similarly appearing diffuse pontine lesions in neonates with spontaneous remissions (Schomerus et al. 2007). In most cases though, the high mortality rate of DIPG has presented a challenge to researchers and clinicians (Bredlau and Korones 2014). Further research in recent years has started to expand the knowledge base about the molecular biology of this tumor hopefully leading to better therapeutic targets in the future.
3.3.2 Pathology
Since the late 1970s, stereotactic biopsy was being performed (Farmer et al. 2001) for patients with DIPG. With the arrival of MR, the characteristic imaging appearance served as a surrogate for diagnosis by tissue confirmation. This position was supported by a report in 1993 from the Children’s Cancer Group recommending against surgical biopsy because of the potential risk (Jones and Baker 2014). Early biopsy results usually demonstrated high-grade glial tumors, although there was variation in grade with a range from WHO grades II to IV (Farmer et al. 2001). Presumably this represented sampling error from the overall tumor, since the majority of these patients demonstrated the relentless phenotype of a high-grade tumor.
3.3.3 Molecular Biology
As techniques have advanced, molecular pathology of diffuse brainstem lesions has been explored further. Only recently a mouse xenograft model has been developed using human high-grade gliomas. Since then, multiple models have been developed including one from a pediatric DIPG biopsy sample (Aoki et al. 2012; Hashizume et al. 2012). In addition, a viable cell line has been developed from a pediatric DIPG biopsy sample (Hashizume et al. 2012). Tissue availability has been limited in the past in the era of imaging-based diagnosis, although rapid autopsy protocols at various institutions have also contributed to tissues available for detailed analyses. A recent review of 300 patients who underwent surgical biopsy showed that stereotactic biopsy could be performed without significant morbidity. The biopsy was aimed either toward clear areas of enhancement on imaging or an area just deep to the cerebellar peduncle in these patients (Cage et al. 2013). In addition, a pediatric neurosurgery consensus conference held in Paris in 2011 agreed that biopsy “was recommended to ascertain biological characteristics to enhance understanding and targeting of treatments, especially in clinical trials” (Walker et al. 2013; Jones and Baker 2014).
Global sequencing and evaluation of gene expression have been performed on samples of high-grade gliomas including diffuse pontine gliomas showing that mutation burdens in DIPG are much higher than in other pediatric cancers. These changes range from structural variants to complicated chromosomal changes caused by chromothripsis (rearrangements with multiple breakpoints that can lead to segments of varying copy numbers) (Kebudi and Cakir 2013).
Among the DIPG tumor samples, more than half the cases have fusion genes detectable by RNA sequencing (Jones and Baker 2014). This is an incidence similar to that of other high-grade gliomas found in the non-brainstem region. Most gene fusions of diffuse brainstem gliomas involved the kinase domain of the three known neurotrophic receptor genes (NTRK).
Growth factor binding to various receptor tyrosine kinases is hypothesized to be a major portion of the oncogenic targets in DIPG. Fusion genes causing activation of the RTK/RAS/PI3K pathway occurred in 69 % of DIPGs. Amplification of platelet-derived growth factor receptor-α (PDGFRA) and c–Met and activation of PI3K have been found in many tumors as well (Grill et al. 2012; Paugh et al. 2011, 2012). This has been of interest given the clinical availability of therapeutic agents that also disrupt PDGF/PDFR ligand activation. Epidermal growth factor receptor (EGFR) has been a subject of interest for inhibition as it is often found as activated in adult high-grade gliomas. In one study, ERBB1 amplification and overexpression were found at an increased amount in escalating grades of brainstem glioma pediatric samples (Gilbertson et al. 2003). In other reports, the activating mutation to produce kinase EGFRvIII expression was rarely observed in DIPG as opposed to other high-grade gliomas.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


