Abstract
This chapter describes the causes, clinical manifestations, diagnosis and treatment of various types of both acquired and inherited bone marrow failure syndromes.
Keywords
Diamond–Blackfan anemia (DBA), Fanconi anemia (FA), dyskeratosis congenita (DC), Shwachman–Diamond syndrome (SDS), amegakaryocytic thrombocytopenia (AMT), congenital dyserythropoietic anemia (CDA), acquired aplastic anemia
Bone marrow failure may manifest as a single cytopenia (e.g., erythroid, myeloid, or megakaryocytic), or as pancytopenia. It may present with a hypoplastic or aplastic marrow or result from invasion of the bone marrow by non-neoplastic (e.g., storage cells) or neoplastic cells.
Bone marrow failure may be congenital or acquired ( Table 8.1 ). Table 8.2 lists the inherited bone marrow failure syndromes (IBMFS) with their causative genes. IBMFS can manifest with pancytopenia (e.g., Fanconi anemia (FA) and dyskeratosis congenita (DC)) or single cytopenias (e.g., Diamond–Blackfan anemia (DBA), Shwachman–Diamond syndrome (SDS), severe congenital neutropenia (SCN), Kostmann syndrome (KS), cyclic neutropenia, amegakaryocytic thrombocytopenia (AMT), and thrombocytopenia absent radii (TAR) syndrome). The “single cell line cytopenias” may develop abnormalities in other hematopoietic cell lines ( Table 8.1 ).
| SINGLE CYTOPENIAS |
|
| TRILINEAGE BONE MARROW FAILURE (GENERALIZED PANCYTOPENIA) |
|
a Can have reduction in other cell lines.
| Disorder | Gene | Number of cases | Locus | Genetics | Gene product |
|---|---|---|---|---|---|
| Fanconi anemia | FANCA | 60–70% | 16q24.3 | Autosomal recessive | FANCA |
| FANCB | ~2% | Xp22.31 | X-linked recessive | FANCB | |
| FANCC | 14% | 9q22.3 | Autosomal recessive | FANCC | |
| FANCD1 | ~3% | 13q12.3 | Autosomal recessive | FANCD1/BRCA2 | |
| FANCD2 | ~3% | 3p25.3 | Autosomal recessive | FANCD2 | |
| FANCE | ~3% | 6p21.3 | Autosomal recessive | FANCE | |
| FANCF | ~2–10% | 11p15 | Autosomal recessive | FANCF | |
| FANCG | ~1% | 9p13 | Autosomal recessive | FANCG/XRCC9 | |
| FANCI | ~2% | 15q25-26 | Autosomal recessive | FANCI/KIAA1794 | |
| FANCJ | <1% | 17q22.3 | Autosomal recessive | FANCJ/BRIP1 | |
| FANCL | <1% | 2p16.1 | Autosomal recessive | FANCL/PHF9/POG | |
| FANCM a | <1% | 14q21.3 | Autosomal recessive | FANCM | |
| FANCN | <1% | 16p12.1 | Autosomal recessive | FANCN/PALB2 | |
| FANCO | <1% | 17q25.1 | Autosomal recessive | FANCO/RAD51C | |
| FANCP | <1% | 16p13.3 | Autosomal recessive | FANCP/SLX4 | |
| FANCQ | <1% | 16p13.12 | Autosomal recessive | FANCQ/XPF/ERCC4 | |
| UBE2T | 1q32.1 | Autosomal recessive | FANCT | ||
| Dyskeratosis congenita | DKC1 | 17–36% | Xq28 | X-linked recessive | Dyskerin |
| TERC | 6–10% | 3q26.2 | Autosomal dominant | TERC/telomerase RNA component | |
| TERT | 1–7% | 5p15.33 | Autosomal dominant and autosomal recessive | TERT/telomerase reverse transcriptase | |
| TINF2 | 11–24% | 14q12 | Autosomal dominant | TIN2 | |
| NOP10 | <1% | 15q14 | Autosomal recessive | NOP10 | |
| NHP2 | <1% | 5q35.3 | Autosomal recessive | NHP2 | |
| WRAP53 | 3% | 17p13.1 | Autosomal recessive | WRAP53 | |
| CTC1 | 1–3% | 17p13.1 | Autosomal recessive | CTC1 | |
| Diamond–Blackfan anemia | RPS19 | 25% | 19q13.2 | Autosomal dominant | RPS19 |
| RPS17 | 1% | 15q25.2 | Autosomal dominant | RPS17 | |
| RPS24 | 2% | 10q22-23 | Autosomal dominant | RPS24 | |
| RPL5 | 7% | 1p22.1 | Autosomal dominant | RPL5 | |
| RPL11 | 5–10% | 1p36.11 | Autosomal dominant | RPL11 | |
| RPL35A | 2–4% | 3q29 | Autosomal dominant | RPL35a | |
| RPS7 | 1% | 2p25.3 | Autosomal dominant | RPS7 | |
| RPS10 | 2–6% | 6p21.31 | Autosomal dominant | RPS10 | |
| RPS26 | 2–6% | 12q13.2 | Autosomal dominant | RPS26 | |
| RPS29 | <1% | 14q21.3 | Autosomal dominant | RPS29 | |
| RPL26 | <1% | 17p13.1 | Autosomal dominant | RPL26 | |
| RPL15 | <1% | 3p24.2 | Autosomal dominant | RPL15 | |
| GATA1 | <1% | Xp11.23 | X-linked recessive | GATA1 | |
| Shwachman–Diamond syndrome (SDS) | SBDS | ~90% | 7q11.21 | Autosomal recessive | SBDS |
| Severe congenital neutropenia (SCN) | ELANE | 60% | 19p13.3 | Autosomal dominant | Neutrophil elastase |
| HAX1 (Kostmann syndrome) | Rare | 1q21.3 | Autosomal recessive | HAX1 | |
| G6PC3 | Rare | 17q21.31 | Autosomal recessive | G6PC3 | |
| GFI1 | Rare | 1p22 | Autosomal dominant | GFI1 | |
| WAS | Rare | Xp11.4-p11.21 | X-linked recessive | WASP | |
| JAGN1 | Rare | 3p25.2 | Autosomal recessive | JAGN1 | |
| GATA2 (MonoMac syndrome) | Rare | 3q21.3 | Autosomal dominant | GATA2 | |
| CXCR4 (WHIM syndrome) | Rare | 2q22.1 | Autosomal dominant | CXCR4 | |
| Amegakaryocytic thrombocytopenia | MPL | 100% | 1p34 | Autosomal recessive | Thrombopoietin receptor/TPOR |
| Thrombocytopenia absent radii (TAR) syndrome | RBM8A | 100% | 1q21.1 | Autosomal recessive | RBM8A |
a FANCM is a member of the “core complex” but homozygosity not yet identified in patients with Fanconi anemia.
Congenital dyserythropoietic anemias (CDAs) result in moderate erythroid failure due to ineffective erythropoiesis with characteristic morphological abnormalities of erythroblasts.
Mitochondrial diseases may present with bone marrow failure (Pearson syndrome, Wolfram syndrome, and various types of sideroblastic anemia).
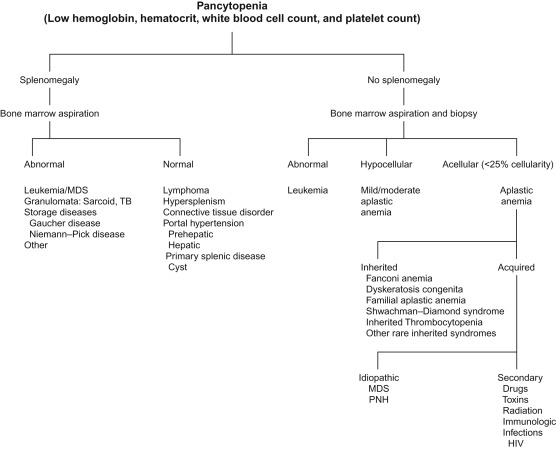
Figure 8.1 shows the differential diagnosis of pancytopenia, and Table 8.3 lists the investigations to be carried out in a patient with pancytopenia.
|
Aplastic Anemia
Aplastic anemia is characterized by a marked decrease or absence of blood-forming elements with resulting pancytopenia and can be inherited or acquired. Various degrees of lymphopenia may be present. Splenomegaly, hepatomegaly, and lymphadenopathy do not generally occur in this condition.
Acquired Aplastic Anemia
Definition
Severe aplastic anemia (SAA) is defined by:
- a.
Bone marrow cellularity of less than 25%.
- b.
At least two of the following cytopenias: granulocyte count <500/mm 3 (<200 mm 3 defines very SAA); platelet count <20,000/mm 3 ; and/or reticulocyte count <20,000/mm 3 .
Non-SAA occurs when the above criteria are not met. There is little consensus on distinguishing between mild and moderate aplastic anemia.
Pathophysiology
Aplastic anemia results from an immunologically mediated, tissue-specific, organ-destructive mechanism. It is postulated that after exposure to an inciting antigen, cells and cytokines of the immune system destroy stem cells in the marrow resulting in pancytopenia. Treatment with immunosuppression leads to marrow recovery.
Gamma-Interferon (γ-IFN) plays a central role in the pathophysiology of aplastic anemia. In vitro studies show that the T-cells from aplastic anemia patients secrete γ-IFN and tumor necrosis factor (TNF). Long-term bone marrow cultures have shown that γ-IFN and TNF are potent inhibitors of both early and late hematopoietic progenitor cells. Both of these cytokines suppress hematopoiesis by their effects on the mitotic cycle and, more importantly, by the mechanism of cell killing. The mechanism of cell killing involves the pathway of apoptosis (i.e., γ-IFN and TNF upregulate each other’s cellular receptors, as well as the Fas receptors in hematopoietic stem cells). Cytotoxic T-cells also secrete interleukin-2 (IL-2), which causes polyclonal expansion of the T-cells. Activation of the Fas receptor on the hematopoietic stem cell by the Fas ligand present on the lymphocytes leads to apoptosis of the targeted hematopoietic progenitor cells. Additionally, γ-IFN mediates its hematopoietic suppressive activity through IFN regulatory factor 1 (IRF-1), which inhibits the transcription of cellular genes and their entry into the cell cycle. γ-IFN also induces the production of nitric oxide, diffusion of which causes additional toxic effects on the hematopoietic progenitor cells. Direct cell–cell interactions between effective lymphocytes and targeted hematopoietic cells probably also occur. The oligoclonal expansion of CD4+ and CD8+ T-cells that fluctuate with disease activity further supports an immune etiology.
Table 8.1 lists the various causes of acquired aplastic anemia.
Clinical Manifestations
Acquired aplastic anemia may be idiopathic or secondary. At least 70% of cases are idiopathic. The incidence is approximately two cases per million per year in the West and higher in parts of Asia (~4–7.5 cases per million per year) and the male:female ratio is 1:1. The onset of acquired aplastic anemia is usually gradual, and the symptoms are related to the pancytopenia:
- •
Anemia results in pallor, easy fatigability, weakness, and loss of appetite.
- •
Thrombocytopenia leads to petechiae, easy bruising, severe nosebleeds and gastrointestinal bleeding and hematuria.
- •
Leukopenia leads to increased susceptibility to infections and oral ulcerations and gingivitis that respond poorly to antibiotic therapy.
- •
Hepatosplenomegaly and lymphadenopathy do not occur and their presence suggests an underlying malignant process.
Laboratory Investigations
- •
Anemia : normocytic or macrocytic, normochromic.
- •
Reticulocytopenia : absolute count more reliable.
- •
Leukopenia : granulocytopenia often less than 1500/mm 3 .
- •
Thrombocytopenia : platelets often less than 30,000/mm 3 .
- •
Fetal hemoglobin : may be slightly to moderately elevated.
- •
Bone marrow :
- •
Marked depression or absence of hematopoietic cells and replacement by fatty tissue containing reticulum cells, lymphocytes, plasma cells, and usually tissue mast cells.
- •
Megaloblastic changes and other features indicative of dyserythropoiesis frequently seen in the erythroid precursors.
- •
Bone marrow biopsy essential to assess cellularity for diagnosis and to exclude the possibility of poor aspiration technique or poor bone marrow sampling; additionally it will help to rule out granulomas, myelofibrosis, or leukemia.
- •
Chromosomal analysis is normal and assists in excluding myelodysplastic syndromes (MDSs).
- •
Bone marrow cultures, antigen-based evaluation, and molecular testing for infectious agents and/or DNA; when indicated.
- •
- •
Chromosome breakage assay : performed on peripheral blood to rule out FA.
- •
Flow cytometry (CD59) : to exclude paroxysmal nocturnal hemoglobinuria.
- •
Telomere length : to screen for DC.
- •
Liver function chemistries : to exclude hepatitis.
- •
Renal function chemistries : to exclude renal disease.
- •
Viral serology testing : hepatitis A, B, and C antibody panel; Epstein–Barr virus (EBV) antibody panel; parvovirus B19 IgG and IgM antibodies; varicella antibody titer; cytomegalovirus (CMV) antibody titer; human immunodeficiency virus antibody test.
- •
Quantitative immunoglobulins (Igs) to rule out immunodeficiency.
- •
Autoimmune disease evaluation : antinuclear antibody (ANA), total hemolytic complement (CH50), C3, C4, direct antiglobulin test (DAT).
- •
HLA typing : patient and family done at the diagnosis of SAA to identify a suitable donor and ensure a timely transplant.
Physical examination, appropriate laboratory screening assays and imaging studies, and, if warranted, mutation analysis should be performed to rule out other IBMFS (DC, DBA, SDS, AMT).
Table 8.4 shows the recommendations for the treatment of moderate and SAA.
|
Treatment
Supportive Care
- •
Transfusion of red cells and platelets should be minimized, but should not be withheld if clearly indicated. The risk of serious bleeding and symptomatic anemia must be balanced against the risk of transfusion sensitization and iron overload.
- •
Prior to any transfusion, perform complete blood group typing to minimize the risk of sensitization to minor blood group antigens and to permit identification of antibodies should they subsequently develop.
- •
Transfusions should be restricted, if possible, to unrelated donors to decrease the likelihood of sensitization to donor antigens.
- •
In all patients, blood products should be leukocyte-depleted to reduce the risk of sensitization and CMV infection. CMV-negative blood products are equivalent to CMV-safe blood products, even for CMV-seronegative patients who may require future transplant.
- •
Patients receiving chronic red cell transfusion should be followed for evidence of iron overload and receive appropriate chelation.
- •
The use of single donor platelets, when available, is recommended.
- •
- •
Menses should be suppressed by the use of contraceptives.
- •
Drugs that impair platelet function, such as aspirin, should be avoided.
- •
Intramuscular injections should be given carefully, followed by ice pack application to injection sites.
- •
The antifibrinolytic agent, ε-aminocaproic acid (Amicar) (100 mg/kg/dose every 6 h, daily maximum 24 grams) can be used to reduce mucosal bleeding in thrombocytopenic patients with good hepatic and renal function. Hematuria is a contraindication to its use. Teeth should be brushed with a cloth or soft toothbrush to avoid gum bleeding.
- •
Avoid infection. Keep patients out of the hospital as much possible. Good dental care is important. Rectal temperatures should not be taken, and the rectal areas should be kept clean and free of fissures. If a patient is febrile:
- •
Culture possible sources, including blood, sputum, urine, stool, skin, and sometimes spinal fluid and bone marrow, for aerobes, anaerobes, fungi, and tubercle bacilli.
- •
Patients with fever and neutropenia should be treated with broad-spectrum antibiotic coverage ( Chapter 33 ). The specific therapy depends upon the clinical status of the patient, the presence of indwelling vascular access devices and knowledge of the local flora pending specific culture results and antibiotic sensitivities. Patients who remain febrile for 4–7 days, even with broad antibacterial coverage, should be started on antifungal therapy empirically. Therapy should be continued until the patient is afebrile and cultures are negative or a specific organism is identified.
- •
- •
Patients previously treated with immunosuppressive therapy (IST) should receive irradiated cellular blood products to prevent transfusion-acquired graft-versus-host disease (GVHD) ( Chapter 36 ). Patients receiving IST should also receive Pneumocystis jiroveci prophylaxis with trimethoprim/sulfamethoxazole (Bactrim/Septra) or pentamidine.
Patients with mild to moderate aplastic anemia should be observed for spontaneous improvement or complete resolution. Hematopoietic stem cell transplantation (HSCT) is the treatment of choice for SAA for patients who have an HLA-matched related donor.
Specific Therapy
Hematopoietic Stem Cell Transplantation
HLA typing should be performed as soon as the diagnosis of SAA is suspected in children. Patients with related histocompatible donors should have an HSCT (FA, DC, paroxysmal nocturnal hemoglobinuria (PNH) or other IBMFSs should be ruled out prior to HSCT). Rapidly treating with HSCT is critical as prolonged neutropenia and multiple transfusions increase the risk of transplant-related morbidity and mortality. See Chapter 34 for preparatory regimens employed pre-transplant.
Immunosuppressive Therapy
Patients unable to undergo matched related HSCT (because no suitable donor is present) should receive IST consisting of antithymocyte globulin (ATG) and cyclosporine (CSA) ( Table 8.5 ). Methylprednisolone or prednisone should be used to prevent serum sickness. The response rate using this regimen is 85%.
|
Contraindications to the use of immunosuppressive drugs include:
- •
Serum creatinine, >2 mg/dl
- •
Concurrent pregnancy.
- •
Concurrent hepatic, renal, cardiac, or metabolic problems of such severity that death is likely to occur within 7–10 days.
- •
Moribund patients.
Antithymocyte Globulin
Test dose : To identify those at greatest risk of systemic anaphylaxis, skin testing is strongly recommended by the manufacturer prior to ATG treatment. An intradermal ATG test dose consisting of 0.02 ml of a 1:1000 dilution in 0.9% sodium chloride solution for injection (5 µg equine IgG) is used with a saline control injection administered on the contralateral side. The results are read at 10 min: a wheal at the ATG site 3 mm or larger in diameter than that at the saline control site suggests clinical sensitivity and increased possibility of a systemic allergic reaction should the drug be dosed intravenously. Many centers have eliminated the test dose in favor of premedication.
The dose of ATG is shown in Table 8.5 .
Usual adverse reactions to ATG :
- •
Thrombocytopenia: Patients may require daily platelet transfusions to maintain a platelet count of more than 20,000/mm 3 (during administration of ATG). Only irradiated and leukocyte-filtered cellular blood products should be used.
- •
Headache.
- •
Myalgia.
- •
Arthralgia.
- •
Chills and fever: Treatment with an antipyretic, an antihistamine, and corticosteroid is indicated as premedication.
- •
Chemical phlebitis: A central line (high flow vein) for infusion of ATG should be used and peripheral veins should be avoided.
- •
Itching and erythema: Treatment with an antihistamine with or without corticosteroids is indicated.
- •
Leukopenia.
- •
Serum sickness may occur approximately 7–10 days following ATG administration. This should be treated by increasing the daily dose of Solumedrol until the symptoms abate.
Uncommon adverse reactions to ATG : Dyspnea, chest, back and flank pain, diarrhea, nausea, vomiting, hypertension, herpes simplex infection, stomatitis, laryngospasm, anaphylaxis, tachycardia, edema, localized infection, malaise, seizures, gastrointestinal bleeding/perforation, thrombophlebitis, lymphadenopathy, hepatosplenomegaly, renal function impairment, liver function abnormalities, myocarditis, and congestive heart failure.
Cyclosporine (CSA) Preparations:
Cyclosporine modified:
- −
Capsules, soft gelatin 50 mg.
- −
Gengraf:
- −
Capsules 25 mg (as cyclosporine modified).
- −
Capsules 100 mg (as cyclosporine modified).
- −
Solution, oral 100 mg/ml (as cyclosporine modified).
- −
Neoral:
- −
Capsules, soft gelatin 25 mg (as cyclosporine modified).
- −
Capsules, soft gelatin 100 mg (as cyclosporine modified).
- −
Solution, oral 100 mg/ml (as cyclosporine modified).
- −
Sandimmune:
- −
Capsules, soft gelatin 25 mg.
- −
Capsules, soft gelatin 100 mg.
- −
Solution, oral 100 mg/ml.
- −
Administer capsules and oral solution on a consistent schedule with respect to time of day and meals.
Gengraf and Neoral oral solution may be diluted with orange juice or apple juice to make the solution more palatable. Sandimmune oral solution may be diluted with milk, chocolate milk, or orange juice, preferably at room temperature, to make it more palatable. Stir mixture well and administer immediately after mixing. Do not allow mixture to stand before administering.
Sandimmune is not bioequivalent to Neoral or Gengraf. Conversion using a 1:1 ratio may result in lower blood concentrations.
The starting dose of cyclosporine is 10 mg/kg per day. CSA levels should be performed once a week for the first 2 weeks and then once every 2 weeks for the remainder of the treatment or as necessary to maintain a whole-blood CSA level between 200 and 400 ng/ml. An elevated serum creatinine level is the principal criterion for dose change. An increase in creatinine level of more than 30% above baseline warrants a reduction in the dose of CSA by 2 mg/kg/day each week until the creatinine level has returned to normal. A serum CSA level of less than 100 ng/ml may be evidence of inadequate absorption and/or non compliance; a CSA level above 500 ng/ml is considered an excessive dose and CSA should be discontinued. Levels should be repeated daily or every other day. When the level returns to 200 ng/ml or less, CSA should be resumed at a 20% reduced dose. In responders, CSA should be tapered very slowly, beginning at 6 months to a year from initiation of therapy although there is little to no evidence available for guidance regarding the target levels and tapering schedule.
Principal side effects of CSA : Renal dysfunction, tremor, hirsutism, hypertension, and gingival hyperplasia.
Uncommon side effects of CSA : Significant hyperkalemia, hyperuricemia, hypomagnesemia, hepatotoxicity, lipemia, central nervous system toxicity (including seizures), and gynecomastia. An increase of more than 100% in the bilirubin level or of liver enzymes is treated in the same way as an increase of more than 30% in creatinine and warrants a reduction in the dose of CSA by 2 mg/kg/day each week until the bilirubin and/or liver enzymes return to the normal range.
Contraindications to the use of CSA : Hypersensitivity to CSA.
Pharmacokinetic interactions with CSA :
- •
Carbamazepine, phenobarbital, phenytoin, rifampin—decreases half-life and blood levels of CSA.
- •
Sulfamethoxazole/trimethoprim IV—decreases serum levels of CSA.
- •
Erythromycin, fluconazole, ketoconazole, nifedipine—increases blood levels of CSA.
- •
Imipenem-cilastatin—increases blood levels of CSA and central nervous system toxicity.
- •
Methylprednisolone (high dose), prednisolone—increases plasma levels of CSA.
- •
Metoclopramide (Reglan)—increases absorption and increases plasma levels of CSA.
Pharmacologic interactions with CSA :
- •
Aminoglycosides, amphotericin B, nonsteroidal anti-inflammatory drugs, trimethoprim/sulfamethoxazole—nephrotoxicity.
- •
Melphalan, quinolones—nephrotoxicity.
- •
Methylprednisolone—seizures.
- •
Azathioprine, corticosteroids, cyclophosphamide—increases immunosuppression, infections, malignancy.
- •
Verapamil—increases immunosuppression.
- •
Digoxin—elevates digoxin level with toxicity.
- •
Non-depolarizing muscle relaxants—prolongs neuromuscular blockade.
Hematopoietic Growth Factors
Granulocyte colony-stimulating factor, G-CSF (Neupogen), had been used to achieve a more rapid increment in the granulocyte count and theoretically to improve protection from infectious complications by stimulating granulopoiesis. G-CSF added to standard ATG and CSA reduces the rate of early infectious episodes and days of hospitalization in very SAA patients but has no effect on overall survival, event-free survival, remission, relapse rates or mortality.
Treatment Choices and Long-Term Follow-Up
Although the short-term outcome with IST is comparable to that obtained with HLA-matched related HSCT, the decision to choose HSCT for younger patients with a histocompatible donor is based on the result of long-term follow-up. There are low rates of late mortality (due to chronic GVHD and therapy-related cancer) in patients undergoing HSCT, and the survival curves are relatively flat. Improved GVHD prophylaxis and safer preparative regimens should further improve these results. In contrast, there is a high risk of clonal hematopoietic disorders (MDS, AML, and PNH) in patients treated with IST compared to HSCT. Those undergoing IST must be closely followed for the development of clonal disorders.
Salvage Therapy
For patients who fail sibling donor HSCT, or have a partial response (ANC ≥500/mm 3 , but are red cell and platelet transfusion dependent) or relapse following IST, management choices include alternative donor HSCT or further IST. HSCT is preferred to IST if a suitable donor is available. Children and teenagers for whom a fully HLA-matched unrelated donor exists (as determined by high-resolution typing) are excellent candidates for an alternative donor HSCT. For patients without a good alternative donor, a second course of ATG/CSA is warranted.
Long-Term Sequelae and Outcomes for SAA
Outcomes for both IST and HSCT have improved considerably in recent years. The results of multiple cohorts report a slightly different response rate and incidence of the clonal evolution of SAA to MDS, AML or PNH. These data vary based on length of follow-up, age of patient, as well as institution/consortium.
- •
Complete or partial response rates in the range of 60–70%, largely from studies in adults, have been reported with IST. Horse ATG appears to be superior to rabbit ATG in these studies. Although the outcomes in children for IST are generally superior to those described for adults, disease-free survival for matched related HSCT is ~95%.
- •
IST improves hematopoiesis and achieves transfusion independence in the majority of patients, but the time to response is long. Hematopoietic response may be partial and relapses are relatively common.
- •
The incidence of clonal hematopoietic disorders including PNH, myelodysplasia, and leukemia in patients with SAA treated with IST ranges from 10–40%. The European Bone Marrow Transplantation Working Party compared the rate of secondary malignancies following HSCT and IST. Forty-two malignancies developed in 860 patients receiving IST, compared to nine in 748 patients who underwent HSCT. In this study, acute leukemia and myelodysplasia were seen exclusively in IST-treated patients while the incidence of solid tumors was similar in the two groups of patients.
- •
From the aggregate data there are a number of conclusions:
- •
Matched sibling donor HSCT is always superior as primary therapy in young patients (<20 years of age) at any neutrophil count.
- •
Immunosuppression, due to transplant-related morbidity and mortality in older patients, is superior to HSCT in older patients (41–50 years).
- •
For the 21–40-year-old age group, the differences are less clear.
- •
In all age groups there are a higher percentage of late failures and clonal evolution in the immunosuppression-treated patients.
- •
When considering the response rate (partial and complete) for IST, the low rate of transplant failure with MUD transplant, the incidence of GVHD and the evolution of clonal disease after IST, the difference in survival between patients treated with MUD donor HSCT and IST increases with time. Thus, MUD transplant (preferably with controlled clinical trials) should be considered as primary therapy for SAA.
- •
Treatment of Moderate Aplastic Anemia
The natural history of moderate aplastic anemia is uncertain and clinical experience varies widely. For this reason, it is generally thought that these patients should be treated initially with supportive therapy with very close follow-up. The majority of patients progress to SAA or develop significant and severe thrombocytopenia and bleeding, serious infections, or a chronic red blood transfusion requirement. These patients should be treated with the same treatment options as described for SAA.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


