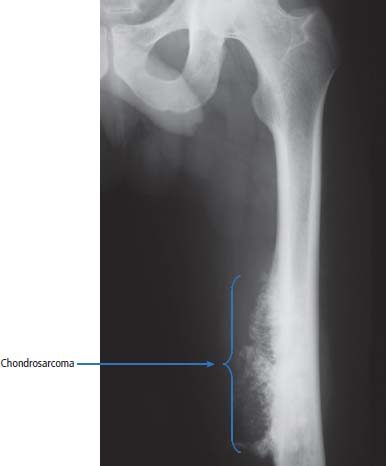
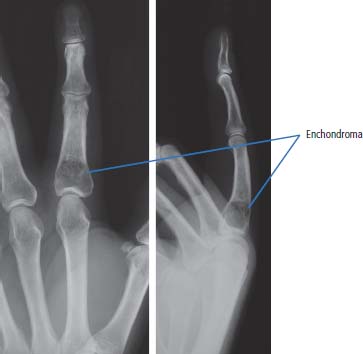
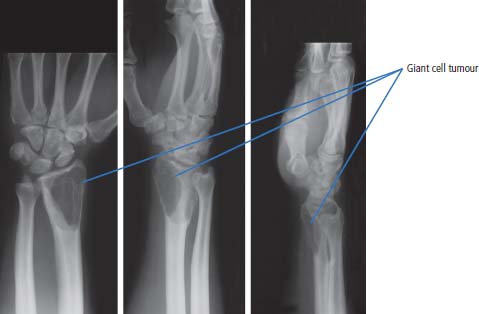
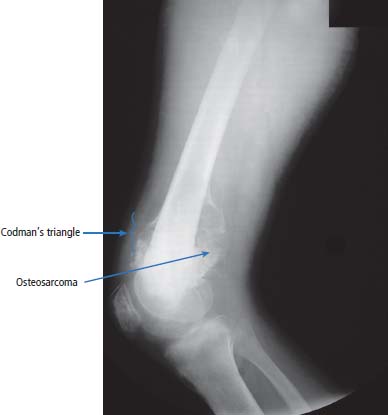
36 Bone tumours are amongst the oldest cancers discovered in humans according to palaeopathological evidence. A Bronze Age woman with bone metastases in her skull has been dated to 1600–1900 BC, whilst Saxon bones from Standlake in Oxfordshire, the United Kingdom, demonstrate features of osteosarcoma in a young adult warrior. St Peregrine, born in 1260 at Forlì, Italy, is the patron saint of cancer sufferers (the feast day is on 4 May). He was due for an amputation for a sarcoma of the leg, but the cancer was cured on the night prior to surgery, following a vision of Christ. He lived a further 20 years and was canonized in 1726. Sarcomas are tumours of the connective tissues or mesenchymal cells, which support the body and include bone, muscle, tendon, fat and synovial tissue. Sarcomas are usually divided into bone tumours and soft tissue sarcomas, although some cancers have feet in both camps, such as Ewing’s sarcoma which may involve either bone or soft tissues. Sarcomas constitute about 1% of adult tumours and there are as many as 50 different subtypes. The presumed cell of origin of the sarcoma defines the classification (Tables 36.1 and 36.2). In 2010, 559 people were diagnosed with bone sarcoma and 3272 with soft tissue sarcoma (Table 36.3). The most common bone tumours are osteosarcoma (35% of primary bone tumours) and Ewing’s sarcoma (20% of primary bone tumours). The most frequent soft tissue sarcomas are leiomyosarcoma (18% soft tissue sarcomas), fibroblastic sarcomas (14%) and liposarcoma (13%), although the largest group of all are the soft tissue sarcoma “not otherwise specified” (20%)! The epidemiology of the different sarcomas varies. For example, osteosarcomas have a bimodal age distribution with peaks in adolescence and in the over-65-year olds, whilst chondrosarcoma incidence rises steadily after the age of 40 years. Ionizing radiation exposure increases the risk of developing bone sarcomas, including therapeutic radiotherapy. Thus there is an increased risk of second primary bone sarcoma in cancer survivors, although occasionally this may be a consequence of a shared inherited genetic predisposition. Thus familial hereditary retinoblastoma increases the risk of not only retinoblastoma but also osteosarcoma and leiomyosarcomas. Similarly in people with Li–Fraumeni syndrome there is an increased risk of osteosarcoma and rhabdomyosarcomas and in those with hereditary multiple exostoses, a high risk of chondrosarcoma. Non-hereditary medical conditions also increase the risk of bone sarcomas. Paget’s disease, which may affect up to 5% of the population, carries a risk of osteosarcoma, although the risk is small (about 1%). Similarly Ollier’s disease is associated with an increased risk of chrondrosarcoma. Kaposi’s sarcoma, both the immunosuppression-associated and classical forms are caused by a human herpesvirus, HHV8/KSHV. Finally, Stewart–Treves syndrome is the rare occurrence of angiosarcoma, developing in a limb affected by chronic lymphoedema, including the arm of women treated for ipsilateral breast cancer. Table 36.1 Origins of primary bone tumours Advances in the molecular biology of sarcomas have resulted in the identification of signature somatic mutations, including chromosomal translocations in subtypes of sarcomas. For example, molecular analysis of Ewing’s sarcoma has revealed a specific chromosomal translocation between chromosomes 22 and 11. This translocation is present in a group of small, round, blue cell tumours which include peripheral neuroectodermal tumours (PNETs), classic Ewing’s and extraosseous Ewing’s sarcoma; these are now grouped together for treatment purposes. In patients with this group of tumours there may be difficulty in obtaining a histopathological diagnosis. Modern advances in molecular biology have led to the identification of the EWS/FLI-1 translocation present in patients with Ewing’s sarcoma. The presence of this translocation is identifiable by fluorescence in situ hybridization (FISH) and this aids diagnosis. Table 36.2 Clinical features of soft tissue sarcomas Table 36.3 UK registrations for sarcomas cancer 2010 Most soft tissue sarcomas occur in the limbs and patients present to their GPs with localized swelling. Patients with bone and soft tissue sarcomas generally present with pain, and the diagnosis usually comes as a result of the classic X-ray appearances of these tumours. Fractures are common and nerve palsies may be seen where there is a cranial presentation. Patients may also present with metastases. Because of the rarity of these tumours and the requirement for a multidisciplinary specialist approach, patients with a suspected diagnosis of sarcoma should be referred on to specialist centres, where results have been shown to be vastly superior to those achieved by peripheral clinics. These tumours are usually diagnosed after a significant delay which ranges from 3 to 6 months from the development of symptoms to diagnosis. Tables 36.4–36.6 list the clinical features of the different types of bone cancers and sarcomas, some of which are illustrated in Figures 36.1, 36.2, 36.3, 36.4 and 36.5. Table 36.4 Clinical features of cartilage-derived bone tumours Table 36.5 Clinical features of osteoid-derived bone tumours Table 36.6 Clinical features of bone tumours of uncertain origins In a patient where a diagnosis of soft tissue sarcoma is suspected, an initial biopsy should be carried out by the surgeon who is to perform definitive surgery. Fine-needle aspiration cytology, core needle biopsy and incisional biopsies are all techniques that are considered by the surgeon, and for those patients with rare abdominal or thoracic soft tissue sarcomas, CT-guided biopsies may be required. The surface entry point of the biopsy needle is tattooed to allow the biopsy tract to be identified and excised at the time of definitive surgery. After the pathological diagnosis has been established, definitive surgery can be planned. This requires a multidisciplinary approach that takes place in the context of magnetic resonance (MR) staging of the local tumour and CT definition of the metastatic sites. The surgical approach requires the removal of the muscle compartment to include the fascia. This limits the risk of local relapse. Figure 36.1 Femur chondrosarcoma showing an expansile lesion with sclerotic margin, cortical destruction and punctuate internal calcification and an associated soft tissue mass. These tumours are most common in middle age and occur around the knee, shoulder or pelvis. Figure 36.2 Enchondroma of the ring finger proximal phalynx showing well-defined lucency and a thin sclerotic rim with preserved cortex. These cartilage-derived tumours occur in 10–50-year-olds most frequently in the diaphyses of the hand or wrist. Multiple enchondromas occur in Ollier’s disease, a non-hereditary condition that is associated with an increased risk of chondrosarcoma. In those patients with Ewing’s sarcoma and osteosarcoma, initial staging will include CT assessment of the chest, abdomen and pelvis, and MR imaging of the primary tumour site. The initial management option for Ewing’s sarcoma includes the consideration of either primary surgery or radiotherapy to control the local lesion. If the lesion is small and it is possible to have substantial resection margins, surgery is the best option with immediate endoprosthetic replacement. For the majority of patients, however, radiotherapy remains the most important treatment modality for the control of local disease. Figure 36.3 Giant cell tumour of the distal radius showing expansion and lucency with cortical destruction giving a multiloculated appearance. These tumours occur most commonly in 20–40-year-olds in long bones at the epiphyses and metaphyses after closure. Figure 36.4 Osteosarcoma of the distal femur showing an expansile soft tissue mass with internal calcification and cortical destruction. There is a marked periosteal reaction with lifting of the periosteum that is described as Codman’s triangle, which is almost always due to an aggressive malignant bone tumour extending into adjacent soft tissues. Figure 36.5 Ewing’s sarcoma of proximal left humerus on (a) MRI and (b) isotope bone scan. Osteosarcomas are rare and for this reason also best managed in specialist centres. This is particularly important for teenage patients with sarcomas. For these patients, chemotherapy, radiation, surgery and counselling all have a significant role in management. Patients with osteosarcomas are generally managed well because of the excellent results achieved using multidisciplinary specialist approaches. In osteosarcoma, bone scanning as well as CT and MR scanning are essential in the initial work-up of a patient. Biopsy of the tumour is required with the open approach preferred. Surgical advances have meant that bone tumours are managed much better than they were, with the aim of limb-sparing prosthetic surgery. For patients with Ewing’s tumours, the last 20 or 30 years have seen a significant evolution of treatment protocols. One type of management generally consists of treatment with induction chemotherapy, followed by local treatment to the primary site with either surgery or radiotherapy or both. This will be followed by further consolidation chemotherapy. Similarly in sarcomas, primary chemotherapy to debulk the tumour is followed by surgery. Both chemotherapy and surgery are complex and highly specialized, requiring immense technical skill and input from many areas of medical and paramedical expertise. Patients with metastatic osteosarcoma can be cured, and, once more, surgery is enormously important. Surgical excision of pulmonary metastases is considered and may be curative in a limited number of patients. The most helpful classification of soft tissue sarcomas is into tumours of fibrous tissue, adipose tissue tumours, tumours of muscle, tumours of blood vessels, tumours of lymph vessels, tumours of synovium, tumours of peripheral nerves, tumours of cartilage and bone-forming tissue, tumours of pleuripotential mesochyme, tumours of uncertain histogenesis and unclassified soft tissue tumours. This latter tumour group is extremely diverse, with at least 50 different subtypes. A simplified classification of the more common adult soft tissue sarcomas is shown in Table 36.7. These groups may in turn be divided into benign and malignant conditions. Benign tumours do not generally metastasize and microscopic examination shows a low mitotic rate. Malignant tumours have a high mitotic rate and do tend to metastasize. Approximately one-third of tumours are low grade and two-thirds are high grade. The clinical features of soft tissue sarcomas are listed in Table 36.2, along with the primary therapy. Table 36.7 Tissue origins and common sites of soft tissue sarcomas There is considerable discussion as to the appropriate management of a soft tissue sarcoma. Low-grade tumours, which by definition should not spread, should be treated by surgical excision alone. Local control should result in 85–100% recovery in these patients. The situation is different for those patients with high-grade tumours, and there is debate as to whether surgery alone, surgery combined with radiation, or surgery, radiation and chemotherapy in combination is the correct approach. There is little argument that surgery is necessary, and the operation of first choice should be one that allows a reasonably wide margin of normal tissue to be excised with the tumour. If a “good” procedure is carried out, such as muscle compartmental excision, the local failure rate is 7–18%. If less radical procedures such as excision biopsy are performed, then the local failure rate is approximately 50%. More radical procedures such as amputation have a lower local recurrence rate of approximately 5%. Over the last decade, there has been a trend towards radical compartmental excision with limb-sparing procedures. After definitive surgery has been performed, the need for radiation and chemotherapy is assessed. Radiation is not given for low-grade tumours. In high-grade tumours, radiotherapy to the tumour bed has an advantage in terms of reduced local recurrence rates in extremity lesions where effective dosages can be given without risking vital structures. Local radiation has no effect upon the progression of distant metastases. Because patients with high-grade sarcomas are at great risk from the progression of their cancer to a metastatic state, adjuvant chemotherapy has been investigated in a number of trials. The original studies, which were non-randomized, showed an advantage to combination chemotherapy. This result has not held up and the consensus view now is that adjuvant chemotherapy has no advantage in terms of 5-year survival. This remains very much a subject for debate, however, and in many centres adjuvant chemotherapy is still administered. The treatment of metastatic soft tissue sarcoma requires the use of chemotherapy. The most effective single-agent treatments lead to responses in 15–35% of patients. Attempts are made to capitalize on this by the use of combination chemotherapy programmes. A slight increase in response rates has been found by some groups of clinicians. This supposed advantage is, however, much debated. Many cancer doctors would advocate the administration of single-agent chemotherapy to their patients simply because combination therapies maximize toxicities and do not provide a significant advantage. There have been advances in treatment that have progressed beyond the use of standard cytotoxic chemotherapy drugs. Imatinib is effective in dermatofibrosarcoma protuberans, pazopanib in non-adipocytic soft tissue sarcomas and sunitinib in alveolar soft part sarcoma and extra-skeletal chondrosarcoma. The prognosis of bone and soft tissue sarcomas vary depending on the stage and the tumour subtype. For bone sarcomas, the 5-year overall survival is 56% but varies from 42% in osteosarcoma to 68% in chondrosarcoma. Similarly, but more markedly, for soft tissue sarcomas the overall 5-year survival is 56% but varies from 30% in angiosarcoma to 99% in dermatofibrosarcoma. Case Study: The council worker who slipped.
Bone and soft tissue sarcomas
Epidemiology
Pathogenesis
Origin
Benign tumour
Malignant tumour
Cartilage
Enchondroma
Chondrosarcoma
Osteochondroma
Origin
Benign tumour
Malignant tumour
Chondroblastoma
Bone
Osteoid osteoma
Osteosarcoma
Osteoblastoma
Unknown origin
Giant cell tumour
Ewing’s sarcoma
Malignant fibrous histiocytoma
Tumour
Age (years)
Commonest sites
Primary therapy
5-year survival
Fibrosarcoma
20–50
Thigh, arm, head and neck
Wide excision and adjuvant radiation
90% (well differentiated)
50% (poorly differentiated)
Liposarcoma
40–60
Thigh, head and neck (rarely arise from lipoma)
Wide excision and adjuvant radiation
66% (myxoid)
10% (pleomorphic)
Embryonal rhabdomyosarcoma
0–10
Head and neck, genitourinary (botyroid)
Neoadjuvant chemoradiation and surgery
40%
Alveolar rhabdomyosarcoma
10–20
Thigh
Neoadjuvant chemoradiation and surgery
60%
Pleomorphic rhabdomyosarcoma
40–70
Thigh, upper arm
Wide excision and adjuvant radiation
10%
Synovial sarcoma
20–40
Leg
Wide excision and adjuvant radiation
40%
Angiosarcoma
50–70
Skin, superficial soft tissues
Wide excision and adjuvant radiation
15%
Leiomyosarcoma
45–65
Retroperitoneal, uterine
Wide excision and adjuvant radiation
40%
Percentage of all cancer registrations
Change in ASR (2000–2010)
5-year overall survival
Female
Male
Female
Male
Female
Male
Bone sarcoma
0.1
0.2
+0%
+0%
54%
58%
Soft tissue sarcoma
1
1
+0%
+17%
56%
56%
Presentation
Enchondroma
Osteochondroma (exostosis)
Chondroblastoma
Chondrosarcoma
Age
10–50 years
10–20 years
5–20 years
30–60 years
Site
Hands, wrist
Knee, shoulder, pelvis
Knee, shoulder, ribs
Knee, shoulder, pelvis
Diaphysis
Metaphysis
Epiphysis prior to fusion
Metaphysis or diaphysis
X-ray
Well-defined lucency, thin sclerotic rim, calcification
Eccentric protrusion from bone, calcification
Well-defined lucency, thin sclerotic rim, calcification
Expansile lucency, sclerotic margin, cortical destruction, soft tissue mass
Notes
Ollier’s disease = multiple enchondromas
1% transform to chondrosarcoma
Osteoid osteoma
Osteoblastoma
Osteosarcoma
Age
10–30 years
10–20 years
10–25 years and >60 years
Site
Knee
Vertebra
Knee, shoulder, pelvis
Diaphysis
Metaphysis
Metaphysis
X-ray
<1 cm central lucency, surrounding bone sclerosis, periosteal reaction
Well-defined lucency, sclerotic rim, cortex preserved, calcification
Lytic/sclerotic expansile lesion, wide transition zone, cortical destruction, soft tissue mass, periosteal reaction, calcification
Giant cell tumour
Ewing’s sarcoma
Malignant fibrous histiocytoma
Age
20–40 years
5–15 years
10–20 years and >60 years
Site
Long bones, knee
Knee, shoulder, pelvis
Knee, pelvis, shoulder
Epiphysis and metaphysis post closure
Diaphysis, less often metaphysis
Metaphysis
X-ray
Lucency with ill-defined endosteal margin, cortical destruction, soft tissue mass, eccentric expansion bone/lung metastases
Ill-defined medullary destruction, small areas of new bone formation, periosteal reaction, soft tissue expansion
Cortical destruction, periosteal reaction, soft tissue mass
Investigations and management of bone sarcomas
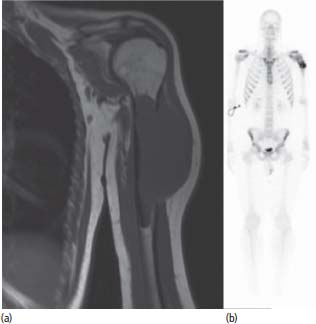
Treatment of Ewing’s sarcomas
Treatment of osteosarcomas
Investigations and management of soft tissue sarcomas
Pathology
Treatment of soft tissue sarcomas
Tissue of origin
Cancer
Usual site in body
Fibrous tissue
Fibroblastic sarcoma
Arms, legs, trunk
Malignant fibrous histiocytoma
Legs
Dermatofibrosarcoma
Trunk
Fat
Liposarcoma
Arms, legs, trunk
Striated muscle
Rhabdomyosarcoma
Arms, legs
Smooth muscle
Leiomyosarcoma
Uterus, GI tract
Blood vessels
Haemangiosarcoma
Arms, legs, trunk
Kaposi’s sarcoma
Legs, trunk
Lymph vessels
Lymphangiosarcoma
Arms
Synovial tissues of joints
Synovial sarcoma
Legs
Peripheral nerves
Malignant peripheral nerve sheath tumour
Arms, legs, trunk
Treatment of the primary tumour
Surgery
Adjuvant chemotherapy and radiation
Treatment of metastatic sarcoma
Prognosis
 ONLINE RESOURCE
ONLINE RESOURCE
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


