Chapter Outline
THE VITAMIN K–DEPENDENT PROTEIN FAMILY
Factor VII (Proconvertin, Convertin)
Factor IX (Plasma Thromboplastin Component, Christmas Factor, Hemophilia B Factor)
Protein C (Autoprothrombin II-A)
FACTOR XI, THE CONTACT PATHWAY CROSSOVER (PLASMA THROMBOPLASTIN ANTECEDENT)
Factor VIII (Antihemophilic Factor, Factor VIII:C)
von Willebrand Factor (Ristocetin Cofactor)
Tissue Factor (Tissue Thromboplastin CD142 and Coagulation Factor III)
THE INTRINSIC (ACCESSORY) PATHWAY FACTORS
Factor XII (Hageman Factor, Contact Factor)
Plasma Prekallikrein (Fletcher Factor)
High–Molecular-Weight Kininogen (Fitzgerald Factor, Williams Factor)
Overview of Coagulation
Generation of the enzyme thrombin from its precursor prothrombin is the central event in the blood coagulation process, essential for hemostasis and the culprit in thrombosis. Blood loss through lack of hemorrhage control has captured the attention of individuals from many walks of life throughout history. At the end of the nineteenth century, the role of thrombin in the clotting of fibrinogen was accurately described by Schmitt. Since that time hemostasis has been synonymous with blood clotting. Congenital diseases associated with absence or reduced production of thrombin (the hemophilias) represent important clinical problems (see Chapters 31 and 32 ). In contrast, the unregulated production of thrombin in an inappropriate location leads to another series of diseases associated with thrombotic occlusion (see Chapter 33 ).
The development of plasma clot–based assays in vitro has played a central role in establishing the logical relationships between the reactants and reactions of hemostasis ( Fig. 27-1 ). The prothrombin time (PT) and the activated partial thromboplastin time (APTT) both rely on the end point of fibrin formation (clotting), which occurs with the generation of less than 5% of the thrombin ultimately produced. The primary pathway leading to hemostatic and thrombotic pathology appears to be associated with the tissue factor (TF)–initiated, “extrinsic” coagulation pathway. In contrast, components unique to the “intrinsic,” or contact, pathway (factor XII, prekallikrein, high–molecular-weight kininogen [HMWK]) do not appear to be essential to hemostasis.
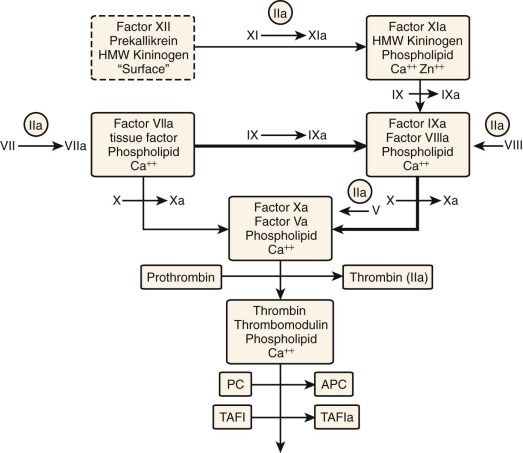
The observation that tissue extracts added to plasma initiated clotting led Quick to develop the PT assay. This assay is still used extensively in the management of warfarin anticoagulant therapy and establishes the continuity of components in the extrinsic pathway . The tissue preparations (thromboplastin) contain the protein TF and phospholipids, with plasma contributing factor VIIa and calcium to form the extrinsic factor Xase , which converts plasma factor X to factor Xa (see Fig. 27-1 ). Factor Xa subsequently combines with plasma factor Va, phospholipids, and calcium to activate prothrombin to thrombin, the latter serving as the catalyst that cleaves fibrinogen to produce fibrin, the clotting end point.
Langdell and colleagues subsequently explored the spontaneous clotting of recalcified citrate plasma and developed the APTT, thus providing the tool that defined the intrinsic pathway of coagulation (see Fig. 27-1 ). This pathway, dependent only on the protein components present in blood plasma, was formalized by the cascade/waterfall hypotheses of MacFarlane and Davie and Ratnoff. The APTT assay involves the addition of an artificial surface, phospholipids, and calcium chloride to blood plasma. Initiation of this contact pathway begins with the surface-dependent activation of factor XII to factor XIIa; this leads to activation of prekallikrein to kallikrein, which, together with HMWK, provides a self-amplifying catalytic system that results in rapid activation of factor XI to factor XIa. Factor XIa, together with HMWK, phospholipid, and metal ions, catalyzes the activation of factor IX to factor IXa. Factor IXa, with factor VIIIa and a membrane surface, form the intrinsic factor Xase , which activates factor X to factor Xa as illustrated in Figure 27-1 .
By the mid-1980s, the conclusion that TF was probably the principal initiator of the hemostatic process and the absence of bleeding associated with deficiencies of the contact pathway initiators (factor XII, prekallikrein, and HMWK) presented a significant conundrum for the scientific community. This was accentuated by the absence of linkage between hemophilia A and B and the extrinsic pathway as evaluated by the PT assay. Subsequently, the intersections between intrinsic and extrinsic and the reactions linking these two “classical” pathways were identified.
Of equal importance to the procoagulant process is its regulation by stoichiometric and dynamic inhibitory systems. Antithrombin (AT) and tissue factor pathway inhibitor (TFPI) are the primary stoichiometric inhibitors, whereas the thrombin-thrombomodulin–protein C system is dynamic in its inhibitory function. The latter catalyst activates protein C to activated protein C (APC), which proteolytically destroys the cofactors factor Va and factor VIIIa, thereby eliminating the intrinsic factor Xase and prothrombinase. TFPI blocks the factor VIIa–TF–factor Xa product complex, thus effectively neutralizing the extrinsic factor Xase ( Fig. 27-2 ). However, TFPI is present in low abundance (≈2.5 nM) in blood and can only delay the hemostatic reaction. AT, normally present in plasma at twice the concentration (≈2 to 3 µM) of any potential coagulation enzyme, neutralizes all the procoagulant serine proteases, primarily in the uncomplexed state.
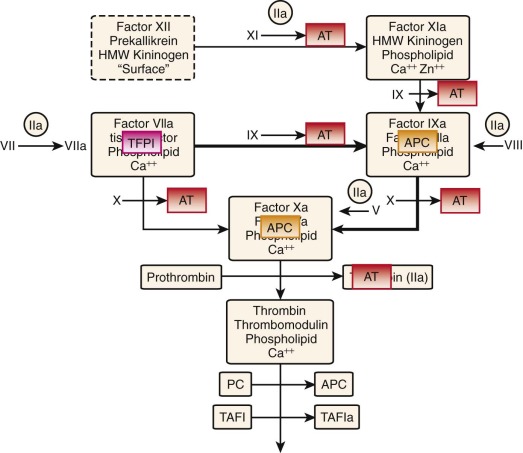
The dynamic protein C system is activated by binding of thrombin to vascular thrombomodulin, with this complex ( Fig. 27-3 ) converting protein C to its activated species APC. APC proteolytically inactivates factors Va and VIIIa. The protein C system, TFPI, and AT cooperate to produce steep TF concentration thresholds that act like a digital “switch” to allow or block thrombin formation.
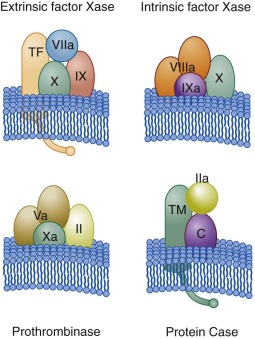
Thus there are three membrane-associated procoagulant complexes (corresponding to the proteases factor IXa, factor Xa, and factor VIIa), each interacting with a cofactor protein (factor VIIIa, factor Va, TF). An analogous anticoagulant complex is composed of thrombin-thrombomodulin (see Fig. 27-3 ). Each complex involves a vitamin K–dependent serine protease, phospholipid, and an associated cofactor protein assembled on a membrane surface provided by an exposed, activated or damaged cell.
The membrane-binding properties of the vitamin K–dependent proteins (VKDPs) are the consequence of posttranslational γ-carboxylation of these macromolecules. The cofactor proteins are either membrane-binding proteins (factor Va, factor VIIIa) recruited from plasma as procofactors or intrinsic membrane proteins (TF, thrombomodulin) regulated by presentation. Each complex catalyst is orders of magnitude (10 3 – to 10 6 -fold) more efficient than the individual serine protease acting on its substrate in solution. When considered on a temporal scale, the amount of thrombin produced by prothrombinase in 1 minute would be produced by factor Xa acting alone in 6 months. As a consequence, factor Xa is ineffective in hemostasis without the presentation of both factor Va and the platelet membrane binding sites on which prothrombinase is formed. The blood coagulation reaction can be described as occurring in three overlapped phases: initiation, propagation, and termination.
Initiation
The factor VIIa–TF–membrane complex (extrinsic factor Xase) ( Fig. 27-4, A ; also see Fig. 27-3 ) catalyzes the activation of both factor IX and factor X, the latter being the more efficient substrate. Thus the initial product formed is factor Xa. Feedback cleavage of factor IX by membrane-bound factor Xa enhances the rate of generation of factor IXa in a cooperative process with the factor VIIa–TF complex.
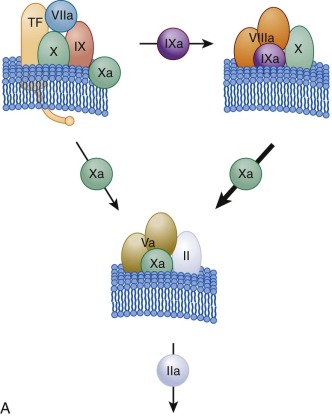
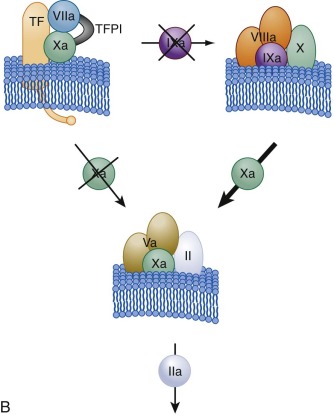
The initially formed, membrane-bound factor Xa activates small amounts of prothrombin to thrombin. This initial activation of prothrombin provides the thrombin essential for acceleration of the hemostatic process by serving as the activator for platelets and the procofactors factor V and factor VIII (see Fig. 27-1 ). Once factor VIIIa is formed, the factor IXa generated by the factor VIIa–TF complex combines with factor VIIIa on the activated platelet membrane to form the intrinsic factor Xase (see Figs. 27-3 and 27-4, A ), which becomes the major activator of factor X. The factor IXa–factor VIIIa complex is 10 6 -fold more active as a factor X activator and 50 times more efficient than the factor VIIa–TF complex in catalyzing activation of factor X (thus the bulk of factor Xa is ultimately produced by the factor IXa–factor VIIIa complex).
Propagation
As the reaction progresses, generation of factor Xa by the more active intrinsic factor Xase exceeds that of the extrinsic factor Xase complex. In addition, the extrinsic factor Xase complex is subject to inhibition by TFPI ( Fig. 27-4, B ). As a consequence, most (>90%) of factor Xa is ultimately produced by the factor VIIIa–factor IXa complex. In hemophilia A and hemophilia B, this amplification of factor Xa generation does not occur. Factor Xa combines with factor Va on the activated platelet membrane receptors, and this factor Va–factor Xa prothrombinase catalyst (see Figs. 27-3 and 27-4 ) converts most prothrombin to thrombin. Prothrombinase is 300,000-fold more active than factor Xa alone in catalyzing activation of prothrombin.
Termination
The same hemostatic process required for preventing leaks from the vasculature may also be life threatening when responsible for intravascular occlusion. Thus equally important to the hemostatic process are the stoichiometric and dynamic inhibitory systems, which block the presentation of thrombin. The sum of inhibitory functions is far in excess of the potential coagulant response. These inhibitory processes governed by TFPI, AT, and the protein C pathway act in synergy to provide activation thresholds, for which a minimum level of TF stimulation must be achieved before significant thrombin generation can occur. The termination phase thus occurs throughout the reaction process.
Flow Regulation
Current data are consistent with the concept of a two-compartment model for regulation of the procoagulant response to a breach in a blood vessel ( Fig. 27-5 ). Regulation of clotting in the extravascular space, apart from providing the initiating TF, is primarily passive and controlled by the availability of fresh blood, whereas regulation in the intravascular compartment is dynamic. Mechanical disruption of a blood vessel results in the movement of blood to an environment where TF is exposed and platelets bind and are activated (see Fig. 27-5 , stage 1 ). These events localize the initiating procoagulant reactions to blood now outside the vasculature, with adhesion and activation of platelets in this extravascular blood providing sufficient receptors and surfaces to support the propagation phase of thrombin generation (see Fig. 27-5 , stage 2 ). This extravascular process rapidly becomes independent of TF as generation of thrombin becomes supplied by the intrinsic factor Xase and prothrombinase complexes. As long as new blood flows into this space, and fibrin formation and platelet aggregation have not achieved a sufficient barrier to stop flow, the system is open and the propagation phase of thrombin generation will continue. If blood ceases to move into the extravascular compartment, the system becomes closed, available prothrombin is consumed, and the complex enzymes are ultimately destroyed or sequestered into inactive complexes (see Fig. 27-5 , stage 3 ). However, the stability of the prothrombin–activating potential incorporated into the platelet-rich thrombus and its localization in the extravascular space ensure that if the barrier springs a leak, robust generation of thrombin will begin immediately on contact and continue until consequent platelet activation and fibrin deposition reestablish a secure barrier.
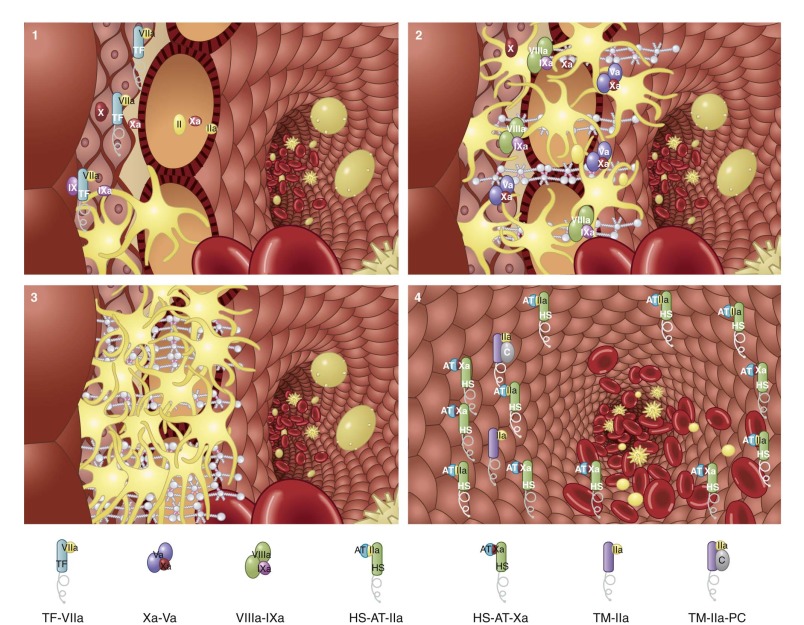
Three mechanisms act to limit clot growth into the intravascular space (see Fig. 27-5 , stage 4 ): thrombomodulin-dependent activation of protein C, the presence of abundant AT, and the actively nonthrombogenic and nonadhesive surface of the vascular endothelium. Thus thrombin generation with consequent ongoing fibrin deposition and platelet activation is suppressed at the extravascular-intravascular boundary by these processes. In contrast, complexes at the extravascular side will remain relatively protected from APC and AT by the forming barrier and are therefore ready to respond to any leakage of blood.
The general properties of proteins of the blood coagulation proteome are summarized in Table 27-1 . It has been stated that “a mouse is not a small furry human.” A corollary is that an infant is not a tiny adult. This is especially true for the average plasma proteome concentration values for the newborn. These data are presented in parentheses in Table 27-1 . Note that except for factor VIII and fibrinogen, most newborn functional protein concentrations are low relative to adult values (see Chapter 5 ).
| Protein | Molecular Weight | PLASMA CONCENTRATION * | Plasma t 1/2 (Days) | Chromosome † (Gene Location) | Functional Classification | |
|---|---|---|---|---|---|---|
| nmol/L | µg/mL | |||||
| INTRINSIC PATHWAY PROTEINS | ||||||
| Factor XII | 80,000 | 500 (265) | 40 (21) | 2-3 | Chr 5 (176 Mb) | Zymogen |
| Factor XI | 160,000 | 30 (11) | 4.8 (2) | 2.5-3.3 | Chr 4 (187 Mb) | Zymogen |
| Prekallikrein | 85,000-88,000 | 486 (180) | 42 (16) | Chr 4 (187 Mb) | Zymogen | |
| HMW kininogen | 120,000 | 670 (362) | 80 (43) | Chr 3 (188 Mb) | Cofactor | |
| LMW kininogen | 66,000 | 1300 | 90 | Chr 3 (188 Mb) | Cofactor | |
| EXTRINSIC PATHWAY PROTEINS | ||||||
| Prothrombin (factor II) | 72,000 | 1400 (672) | 100 (48) | 2.5 | Chr 11 (47 Mb) | VKD zymogen |
| α-Thrombin | 37,000 | Serine protease | ||||
| Factor VII | 50,000 | 10 (7) | 0.5 (0.3) | 0.25 | Chr 13 (113 Mb) | VKD zymogen |
| Factor IX | 55,000 | 90 (48) | 5 (3) | 1 | X Chr (138 Mb) | VKD zymogen |
| Factor X | 59,000 | 170 (68) | 10 (4) | 1.5 | Chr 13 (113 Mb) | VKD zymogen |
| Protein C | 62,000 | 65 (23) | 4 (1) | 0.33 | Chr 2 (128 Mb) | VKD zymogen |
| Protein S | 69,000 | 300 (108) | 20 (7) | 1.75 | Chr 3 (95 Mb) | VKD protein |
| Protein Z | 62,000 | 47 | 3 | 2.5 | Chr 13 (113 Mb) | VKD protein |
| Factor V | 330,000 | 20 (14) | 6.6 (5) | 0.5 | 1q21-q25 | Procofactor |
| Factor VIII | 285,000 | 1.1-1.5 ‡ | 0.3-0.4 ‡ | 0.3-0.5 | X Chr (154 Mb) | Procofactor |
| VWF | 255,000 (monomer) | Varies | 10 | Chr 12 (6 Mb) | Platelet adhesion, factor VIII carrier | |
| Tissue factor | 44,000 | — | — | — | Chr 1 (9 Mb) | Cell-associated cofactor |
| Thrombomodulin | 100,000 | — | — | — | Chr 20 (23 Mb) | Cofactor |
| Fibrinogen | 340,000 | 7400 (8380) | 2500 (2830) | 3-5 | Chr 4 (156 Mb) | Structural protein cell adhesion |
| Aα | 66,500 | |||||
| Bβ | 52,000 | |||||
| γ | 46,500 | |||||
| Factor XIII | 320,000 | 94 (72) | 30 (23) | 9-10 | Transglutaminase zymogen | |
| A chain | 83,200 | Chr 6 (6 Mb) | ||||
| B chain | 79,700 | Chr 1 (195 Mb) | ||||
| Tissue factor pathway inhibitor | 40,000 | 1-4 | 0.1 | 6.4 × 10 −4 – 1.4 × 10 −3 | Chr 2 (188 Mb) | Kunitz inhibitor |
| Antithrombin III | 58,000 | 2400 (1510) | 140 (88) | 2.5-3 | Chr 1 (172 Mb) | Serpin inhibitor |
| Heparin cofactor II | 66,000 | 950 (408) | 62 (26) | 2.5 | Chr 22 (19 Mb) | Serpin inhibitor |
| Protein C inhibitor | 57,000 | 90 | 5 | 1 | Chr 14 (94 Mb) | Serpin inhibitor |
† Chromosome number. Numbers in parentheses are approximate positions in megabases (from human genome build 36.2 www.ncbi.nlm.nih.gov ).
* Concentrations are reported as the mean of the healthy adult population, with the mean of the healthy 1-day-old infant population in parentheses. (Infant values from Andrew M, Paes B, Milner R, et al: Development of the human coagulation system in the full-term infant, Blood 70:165-172, 1987.)
‡ Butenas S, Parhami-Seren B, Mann KG: The influence of von Willebrand factor on factor VIII activity measurements. J Thromb Haemost 7(1):132-137, 2009; Butenas S, Parhami-Seren B, Undas A, et al: The “normal” factor VIII concentration in plasma. Thromb Res 126(2);119-123, 2010.
The Vitamin K–Dependent Protein Family
The Nobel Prize was awarded to Dam and Doisy in 1941 for their discovery of the fat-soluble vitamin K. Nearly simultaneously with the discovery of vitamin K, a naturally occurring antagonist, bishydroxycoumarin (dicumarol), was described. This antagonist was identified as a toxic agent in spoiled sweet clover that caused hemorrhage in cattle because of decreased thrombin formation. This agent was introduced as an anticoagulant in humans in 1941.
Vitamin K, essential for the biosynthesis of clotting factors, contributes to the conversion of 10 to 13 glutamate residues to γ-carboxyglutamate residues (Gla). This posttranslational modification endows the vitamin K–dependent protein (VKDP) with Ca 2+ -binding and membrane-binding qualities. Gla formation is decreased or absent after treatment with coumarin derivatives.
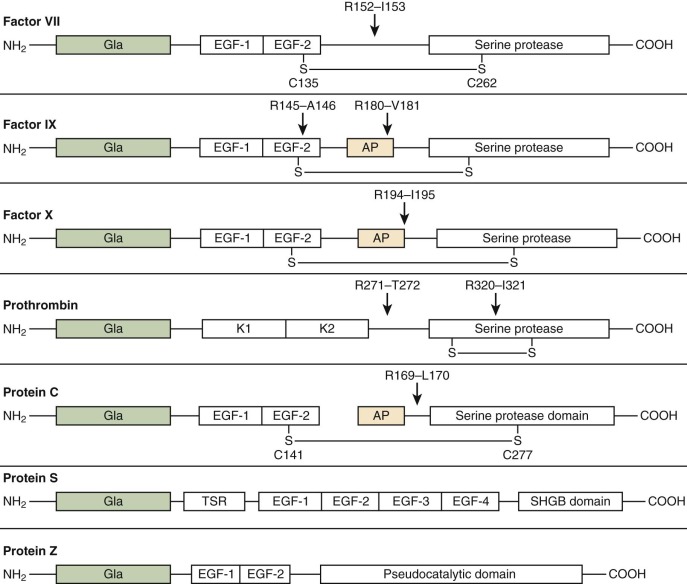
The VKDPs ( Fig. 27-6 ) can be divided into two classes: procoagulant (factors II, VII, IX, and X) and anticoagulant (protein C, protein S, and protein Z). The homology among these proteins is probably due to a common ancestral gene. Each VKDP is composed of discreet domains characterized by highly conserved regions. The NH 2 -terminal “Gla” domains (see Fig. 27-6 ) are followed by either kringle domains in prothrombin or epidermal growth factor (EGF)–like domains in factor VII, factor IX, factor X, protein C, protein S, and protein Z. Protein S contains four EGF domains followed by a sex hormone–binding domain (SHD). The serine protease domain for prothrombin, factor VII, factor IX, factor X, and protein C becomes functional after cleavage of specific peptide bonds. Protein S is not a serine protease precursor but is sensitive to thrombin cleavage. Protein Z contains a “pseudocatalytic domain” in the (COOH)-terminal but cannot function as a serine protease.
Synthesis of these proteins occurs primarily in the liver, although synthesis in other tissue has been reported. The plasma concentration of circulating VKDP varies 200-fold from approximately 100 µg/mL for prothrombin to 0.5 µg/mL for factor VII (see Table 27-1 ). Clearance rates of the VKDPs vary with their half-lives, from 6 hours (factor VII) to 2.5 days (prothrombin; see Table 27-1 ).
The VKDPs are synthesized as preproproteins and are modified posttranslationally at specific glutamates (γ-carboxylation) to form γ-carboxyglutamic acid (Gla). Other modifications include β-hydroxylation at select aspartate and asparagine residues, tyrosine sulfation, and glycosylation. After synthesis of the single-chain precursors, the hydrophobic signal peptide directs each polypeptide to the endoplasmic reticulum. The propeptides, which play a role in docking vitamin K–dependent carboxylase, are removed by an endoproteinase. For factor X and protein C, removal of an internal dipeptide or tripeptide converts them to their mature plasma two-chain zymogen forms.
The Gla domains constitute the first approximately 50 residues of the VKDPs (see Fig. 27-6 ). The negative charge elicited from the string of Gla residues (9 to 13) contributes to the binding of Ca 2+ and generation of the conformation required for binding to anionic phospholipid membranes. In vivo , this surface is supplied by activated platelets or other blood cells in response to vascular damage through exposure of the phosphatidylserine-rich internal face of their cell membranes. Without vitamin K, or in the presence of vitamin K antagonists, the coagulation protein precursors are synthesized but not γ-carboxylated. In this form they are still secreted into plasma but display diminished function. The γ-carboxylation reaction is catalyzed by the enzyme γ-glutamyl carboxylase. This enzyme is located in the rough endoplasmic reticulum and requires the reduced form of vitamin K, oxygen, and carbon dioxide to convert glutamic acid residues to γ-carboxyglutamic acid.
Vitamin K 1 (phylloquinone) is primarily found in leafy green vegetables and vegetable oil. Additional K activity may be provided by vitamin K 2 (menaquinones) synthesized by intestinal gram-negative bacteria. Synthetic vitamin K 3 (menadione) has no intrinsic activity until it undergoes in vivo modification to the active menaquinone form. In vivo, vitamin K is recycled in a microsomal oxidation-reduction system for continued use in the γ-carboxylation reaction. In order to perform the γ-carboxylation reaction, vitamin K has to be present in its reduced hydroquinone form. As the precursor proteins are carboxylated, vitamin K is oxidized to the epoxide. The epoxide in the presence of 2,3-epoxide reductase, with thiols being used as the reducing agent, yields the quinone form of vitamin K. A subsequent reduced nicotinamide adenine dinucleotide phosphate (NADPH) or NADH-dependent quinone reductase reaction resynthesizes the hydroquinone form. The cycle can thus continue. Warfarin anticoagulant therapy blocks the process required for the regeneration of reduced hydroquinone vitamin K. Because warfarin and other coumarin–derivative drugs indirectly affect carboxylation, they can be overcome with excess vitamin K.
The Gla domain is followed by two tandem EGF domains (EGF-1 and EGF-2) in factor VII, factor IX, factor X, and protein C and four EGF domains (EGF-1 to EGF-4) in protein S (see Fig. 27-6 ). Each EGF-like domain consists of 40 to 50 amino acids, including 6 cysteine residues involved in disulfide bond formation. These domains are found widely distributed in extracellular and membrane proteins. Proteins containing these EGF-like domains are involved in blood coagulation, fibrinolysis, complement activation, connective tissue microfibril formation, and signal transduction. The function of the EGF domain is still unclear. One hypothesis is that it serves as a spacer. There is a consistent elongation of the molecules of factors VII, IX, and X, protein C, and protein S. The distance between the membrane-binding Gla domain and the serine protease domain is crucial to placement of the active site in an appropriate position relative to the target peptide bond in its substrate.
The serine protease domains (see Fig. 27-6 ) account for approximately half the mass of each protein. Peptide bond cleavage at specific sites converts the vitamin K–dependent zymogens to their active, chymotrypsin-like, serine protease forms. The mechanism of proteolysis by chymotrypsin involves a catalytic triad composed of Asp102, His57, and Ser195 (chymotrypsin numbering). The serine protease domains of all the Gla-containing factors are homologous members of the chymotrypsin family but cleave almost specifically at arginyl residues. However, unlike trypsin, which shows little specificity beyond the requirement for arginyl or lysyl residues at the cleavage site, the activated coagulation factors have extended substrate specificity pockets whereby only selected amino acid sequences are recognized by each protease. Thus despite the high degree of homology between the protease domains of protein C and factors II, VII, IX, and X, each of these factors has a highly specific function in coagulation. Some of this discrimination is mediated by surface loops, along with other domains distant from the substrate binding pocket termed exosites.
Factor VII (Proconvertin, Convertin)
The single-chain factor VII zymogen is activated to its two-chain protease form by cleavage of a single peptide bond between Arg152 and Ile153 (see Fig. 27-6 ). The mechanism for the initial activation of this zymogen is unclear. The resulting protease consists of an NH 2 -terminal light chain linked by a single disulfide bond to a COOH-terminal heavy chain containing the catalytic domain. Factor VIIa has very poor catalytic efficiency without the presence of its cofactor TF. Cleavage of factor VII to factor VIIa can be catalyzed by several proteases, including α-thrombin, factor IXa, factor Xa, factor XIIa, and factor VII–activating protease, as well as by autoactivation by factor VIIa. The active factor VIIa serine protease bound to TF forms the extrinsic factor Xase complex (see Fig. 27-3 ).
Factor VIIa, unlike other serine proteases of the coagulation cascade, is not readily inhibited by circulating protease inhibitors, including the AT-heparin complex, unless bound to TF. Thus regulation of TF expression is the primary means of controlling factor VIIa activity. Paradoxically, the zymogen factor VII (10 nM) is an effective competitor of factor VIIa (0.1 nM) for binding to TF. This competition downregulates the level of enzymatically active complex (factor VIIa–TF), thereby suppressing initiation of the clotting cascade. Once the extrinsic tenase complex activates factor X to factor Xa, TFPI can form a quaternary complex (factor VIIa–TF–factor Xa–TFPI) with no enzymatic activity (see Fig. 27-4 ). Thus once assembled and functioning, the extrinsic tenase, along with its substrate factor Xa, is a target for inactivation through the action of TFPI.
Factor IX (Plasma Thromboplastin Component, Christmas Factor, Hemophilia B Factor)
Factor IX is a single-chain zymogen consisting of 415 amino acids organized with a Gla domain (12 Gla residues), two tandem EGF domains, an activation peptide region, and a serine protease domain (see Fig. 27-6 ). Glycosylation in the form of O-linked and N-linked oligosaccharides makes up 17% of the factor IX protein by weight. Activation of factor IX is a two-stage process that requires cleavage at Arg145 and Arg180 and results in the release of a 35-residue activation peptide (see Fig. 27-6 ). The physiologic activators of factor IX are either TF–factor VIIa (extrinsic tenase complex) or factor XIa. The first cleavage at Arg145-Ala146 results in factor IXα. This cleavage has been shown to be important in its affinity for its cofactor factor VIIIa. The second cleavage occurs at Arg180-Val181 and results in factor IXαβ, the active form, also referred to as factor IX a. Both cleavages are required for the biologic activity of factor IXa. Plasma factor IXa is primarily inhibited by AT (see Fig. 27-2 ). In vitro experiments conducted on phospholipid vesicles and cell membranes showed that protease nexin-2/amyloid β-protein precursor (PN2/APP) can also inhibit factor IXa.
Factor X (Stuart Factor)
Factor X is synthesized in the liver and circulates as a two-chain molecule linked by a disulfide bond. The NH 2 -terminal light chain consists of a Gla domain and two EGF-like domains (see Fig. 27-6 ). The COOH-terminal heavy chain consists of an activation peptide region and a catalytic (serine protease) domain. Factor X is activated by both the intrinsic and extrinsic factor Xases (see Figs. 27-1 and 27-3 ) by cleavage at Arg194-Ile195 (see Fig. 27-6 ). A 52–amino acid activation peptide is released. The resulting catalytic serine protease, factor Xa, is composed of a light chain and a disulfide-linked heavy chain. Factor X has to be bound to the membrane before activation. Factor Xa participates in forming the prothrombinase complex (see Fig. 27-4 ). The propagation phase of thrombin generation is catalyzed principally by factor Xa generated via the intrinsic tenase complex (see Fig. 27-3 ), which activates factor X at a 50- to 100-fold higher rate than the extrinsic tenase complex does. The factor Xa active site associates with the COOH-terminus of TFPI. The factor Xa–TFPI complex rapidly inactivates TF–factor VIIa to form a stable quaternary complex, TF–factor VIIa–TFPI–factor Xa. Factor Xa is inhibited by AT but only poorly when in the prothrombinase complex.
Factor II (Prothrombin)
Prothrombin is the single-chain precursor of factor IIa (thrombin), the key enzyme in blood coagulation. The zymogen prothrombin is the most abundant of the plasma VKDPs and circulates in plasma at a mean concentration of 1.4 µM, or 100 µg/mL (see Table 27-1 ). Prothrombin is synthesized primarily in the liver. Low levels of prothrombin expression have also been identified in other tissues, including the brain, diaphragm, stomach, kidney, spleen, intestine, uterus, placenta, and adrenal gland. Removal of the 43–amino acid propeptide generates the mature protein of 579 amino acids. An NH 2 -terminal Gla domain is followed by two kringle domains and the serine protease precursor domain (see Fig. 27-6 ). Kringles have been identified as common motifs in many plasma proteins, including prothrombin, plasminogen, tissue plasminogen activator, urokinase, factor XII, and apolipoprotein(a). The kringle 1 domain may interact with factor Va, although kringle 2 appears to be the primary region mediating interaction with factor Va to form the prothrombinase complex. This latter interaction may initiate conformational changes that make reaction sites accessible for enzymatic cleavage of prothrombin. The NH 2 -terminal Gla domain and the first kringle domain together are referred to as prothrombin fragment 1 , with the kringle 2 domain being located in prothrombin fragment 2. The sequence of the enzyme α-thrombin is contained within prethrombin 2.
Activation of prothrombin by prothrombinase proceeds through an initial cleavage of prothrombin at Arg320-Ile321, which gives rise to meizothrombin, a two-chain disulfide-linked molecule (see Fig. 27-6 ). Meizothrombin expresses some of the activities of α-thrombin. However, the fibrinogen clotting ability of meizothrombin is impaired in comparison to that of α-thrombin. Meizothrombin is subsequently cleaved at Arg271-Thr272 to yield the NH 2 -terminal half of the molecule (prothrombin fragment 1.2) and α-thrombin. α-Thrombin consists of an NH 2 -terminal 49-residue A-chain disulfide linked to a COOH-terminal 259-residue B chain containing the catalytic triad.
An alternative cleavage pathway occurs when factor Xa independently acts on prothrombin. In the presence of factor Xa and Ca 2+ , the initial cleavage occurs at Arg271-Thr272 and gives rise to prothrombin fragment 1.2 and prethrombin 2. Subsequent cleavage at Arg320 in prethrombin 2 yields α-thrombin. Prothrombin fragment 1.2 remains noncovalently associated with α-thrombin. In contrast to other VKDPs, the phospholipid-binding Gla region is not covalently attached to the serine protease domain after cleavage of prothrombin to thrombin.
α-Thrombin is the central enzyme in blood coagulation in that it contributes to reactions at all levels, thereby allowing overall maintenance of vascular integrity. Its premier role in hemostasis results in platelet-fibrin clot formation. However, the events that lead up to and beyond fibrin formation also involve protein cleavage by α-thrombin, as well as activating platelets, factor VII, factor V, factor VIII, factor XI, and factor XIII. The contributions of α-thrombin also extend to anticoagulant and fibrinolytic reactions, including activation of protein C and thrombin-activatable fibrinolysis inhibitor (TAFI).
The stable form of human α-thrombin possesses at least five distinct binding sites for substrates, inhibitors, cofactors, apolar molecules, and sodium ions (Na + ). Exosite I, the fibrinogen binding site, is an electropositive anion binding site distinct from, but acting in concert with, the active site of the α-thrombin molecule. In addition to fibrinogen, exosite I also recognizes the COOH-terminal domain of hirudin, the hirudin-like region of PAR-1, and the fifth and sixth EGF-like domains of thrombomodulin.
Many of the effects of α-thrombin on platelets and cells are elicited through the interaction of α-thrombin with receptor molecules, with α-thrombin binding to a receptor and initiating a signal transduction mechanism. The interaction between α-thrombin and the human platelet thrombin receptor PAR-1, however, is characterized by a more unusual mechanism in which the receptor is also a substrate for α-thrombin. α-Thrombin cleaves the extracellular region of the receptor and produces a “tethered ligand.” The new NH 2 -terminus binds back to and activates the receptor. Two additional α-thrombin receptors homologous to PAR-1, PAR-3, and PAR-4 have been identified. α-Thrombin’s role continues beyond hemostasis into the realm of tissue repair and the remodeling phase necessary to regenerate damaged vascular tissue. α-Thrombin is a potent mitogen and stimulates cell division in macrophages, smooth muscle cells, and endothelial cells.
Protein C (Autoprothrombin II-A)
Protein C circulates as a heterodimer consisting of disulfide-linked heavy and light chains (see Fig. 27-6 ). However, approximately 5% to 10% of circulating protein C is in the single-chain form. The NH 2 -terminal light chain contains the Gla domain and a hydrophobic region that connects the Gla domain to two EGF domains. The COOH-terminal heavy chain contains the serine protease domain. Cleavage at Arg169-Leu170 releases the activation peptide from the heavy chain to generate the enzyme. Although the α-thrombin–thrombomodulin complex is probably the major physiologic activator of protein C, other potential activators include plasmin and factor Xa. The half-life of protein C (8 to 10 hours; see Table 27-1 ) is markedly shorter than the half-life of other members of the VKDP family, which is probably the basis of the transient hypercoagulable state that may occur subsequent to the administration of coumarin-based anticoagulants.
The anticoagulant role of the dynamic protein C inhibitory pathway is proteolytic inactivation of factor Va. APC inactivates factor Va via a sequential series of proteolytic cleavages and thus inhibits the generation of α-thrombin. APC also cleaves and inactivates factor VIIIa, although the spontaneous dissociation/inactivation of factor VIIIa is probably the physiologic regulator of factor Xa generation. Complete inactivation of factor Va by APC requires an anionic phospholipid surface, and rates of inactivation of factors Va and VIIIa by APC have been reported to be enhanced by protein S. APC also has profibrinolytic effects associated with decreased activation of TAFI. TAFI is activated by the α-thrombin–thrombomodulin complex and acts to prolong clot lysis. Cleavage of factor Va by APC inhibits α-thrombin generation, thus reducing α-thrombin–thrombomodulin–mediated TAFI activation.
Protein C activation and APC activity are controlled on several levels. The endothelial cell protein C receptor (EPCR) provides cell-specific binding sites for both protein C and APC. Inflammatory agents such as endotoxin, interleukin-1β (IL-1β), tumor necrosis factor-β (TNF-β), and TNF-α upregulate protein C activation on endothelial cells. TNF-α downregulates thrombomodulin on the endothelial cell surface. There are multiple other factors that downregulate thrombomodulin as well. APC activity is further regulated by the protein C inhibitor, plasminogen activator inhibitor 3 (PAI-3).
Protein S
Protein S is a VDKP that is not a serine protease precursor. Approximately 40% of protein S circulates in the free form, and the remaining 60% circulates as a 1 : 1 complex with C4b-binding protein (C4bBP), a regulatory protein of the complement system. The precise function of protein S in the protein C inhibitory pathway is controversial. Protein S is thought to function as a cofactor for APC in the inactivation of factors Va and VIIIa. Protein S has also been reported to bind to factor Xa, factor VIII, and factor Va and compete for prothrombinase binding sites on the membrane surface. The C4bBP–protein S complex may inhibit factor X activation as well. Protein S may have other roles apart from anticoagulation. α-Thrombin cleavage of protein S inhibits the ability of protein S to act as a cofactor for APC. The anticoagulant activity of protein S is also neutralized when bound to C4bBP.
Protein Z
Protein Z is an enzymatically inactive homologue of factors VII, IX, and X and protein C. Protein Z circulates in plasma in a complex with protein Z–dependent protease inhibitor (ZPI). ZPI is a 72-kD member of the serpin superfamily. However, like protein S, protein Z does not function as a protease. Although the COOH-terminus contains a homologue to the catalytic domains present in serine protease zymogens, the catalytic triad is not present. The function of protein Z in vivo is unclear. Thrombosis is observed in mice when factor V Leiden is protein Z deficient.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


