B-Lymphocyte Responses
Michael McHeyzer-Williams
INTRODUCTION
B-lymphocyte responses provide the effective and longlasting immune protection induced by most vaccines in use today. We measure antibody molecules as the circulating agent of immune protection but now understand much more about the underlying B-lymphocyte response that progressively matures in response to foreign antigen exposure. This chapter will focus on the highly-regulated cellular and molecular development of antigen-specific B-lymphocyte responses.
The three cardinal features of effective B-cell immunity are antigen specificity, antibody class, and antigen binding affinity. To be effective, antibodies must bind vulnerable antigens on the targeted pathogen. During infection, this is a struggle between the evasion mechanisms of the pathogen and preexisting diversity within the adaptive immune system. Different classes of antibodies engage distinct mechanisms of antigen clearance. These various classes of antibody provide either more or less protection depending on the pathogen’s portal of entry at infection. Finally, the right antigen-specific antibody still requires induction of sufficiently high binding affinity to provide adequate sensitivity for long-term immune protection. Many promising antigens fail to achieve adequate immunogenicity in contemporary vaccine strategies due to poor affinity maturation. Here, we focus on the sequential mechanisms that program these central attributes of effective antigen-specific B-cell memory.
In the past few years, experimental access to immune response biology has dramatically shifted with the advent of multiphoton laser-based intravital imaging techniques. These studies provide direct access to the mechanics and cell dynamics of antigen-specific cognate regulation in vivo. This information serves to integrate existing knowledge in the field using a real-time scaffold for developmental progression in vivo. Importantly, follicular helper T (TFH) cells have recently emerged as a new class of antigen-specific immune regulator that controls multiple stages of high-affinity B-cell immunity. Understanding antigen-specific TFH-cell development and function remains an active research challenge with great potential for the rational design of future vaccines. Current information regarding the role of antigenspecific TFH cells will be integrated into this chapter to provide a regulatory dimension to B-lymphocyte responses.
Following initial exposure to antigen, TFH -cell-regulated B-cell immunity progresses in three separable stages of antigen-specific development. Each stage is characterized by a B-cell antigen-recognition event followed by contact with cognate TFH cells that determines subsequent B-cell fate. Naïve B cells recognize foreign antigen and then present antigenic peptides to specific TFH cells to progress in development across two major pathways (pre-germinal center [GC] development). Extrafollicular development permits antibody class switch and plasma cell differentiation while entry into the GC reaction is the major pathway to highaffinity B-cell memory (GC cycle). GC B cells in this pathway can switch antibody class and affinity mature their expressed B-cell receptor (BCR) following access to antigen and presentation to GC TFH cells. Upon antigen reexposure, memory B cells recognize, uptake, and then present antigen to memory TFH cells to promote rapid memory B-cell responses and boost circulating high-affinity antibody (memory B-cell response). Antigen recall is the least studied facet of B-lymphocyte responses but provides an important developmental juncture for vaccine-based prophylactic or therapeutic intervention.
This chapter mainly focuses on what is known of antigen-specific B-lymphocyte responses in mouse models with reference to work conducted in humans. Further, there is an emphasis on the response to model antigens that provides a greater understanding of how to manipulate adaptive immunity for preventative vaccination rather than a focus on the immune response to infection.
PRE-GERMINAL CENTER DEVELOPMENT
The initial antibody response to many infectious agents is largely based on the rapid T-cell independent (TI) expansion of B cells and their subsequent differentiation into plasma cells. B1 and marginal zone B cells are largely responsible for these rapid TI humoral responses suggesting that some level of “natural memory” function resides in these B-cell subsets and may be predetermined in an evolutionarily conserved manner. TI antigens can be separated into two broad categories based on their ability to polyclonally activate B cells (TI-1) or require BCR recognition of multivalent epitopes to induce B-cell differentiation in the absence of T-cell help (TI-2). TI-2 antigens can activate BCR signaling but require accessory signals to promote the development of antigen-specific plasma cells. In contrast, monovalent protein antigens require antigen-specific helper T-cell regulation to promote high titer antibody responses and the development of B-cell memory. These TFH-cell-dependent antibody responses take longer to emerge and display a spectrum of BCR affinities and antibody isotypes as multiple strategies for antigen-specific clearance in vivo. Immune responses to
model protein antigens provide experimental access to this complex cascade of cellular and molecular events that underpin long-term protective immunity.
model protein antigens provide experimental access to this complex cascade of cellular and molecular events that underpin long-term protective immunity.
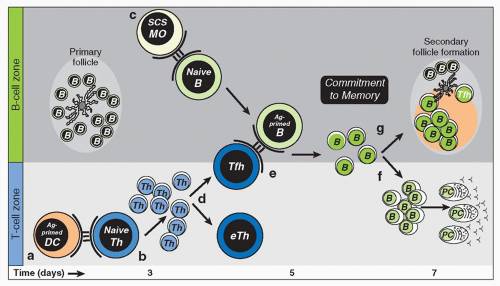 FIG. 10.1. Pre—Germinal Center (GC) Development. A: Local protein vaccination induces dendritic cells (DC) maturation and migration to the T-cell zones of draining lymph nodes (LNs). B: Peptide major histocompatibility class II expressing DCs will engage naive antigen-specific helper T (TH) cells to induce proliferation and effector TH-cell differentiation. C: Whole antigen will be trapped by subcapsular sinus macrophage and presented to naive follicular B cells. Antigen-specific B cells will become activated and uptake, process, and present antigenic peptides and migrate toward the T-B borders of the draining LN. D: Effector TH cells emerge in multiple forms with emigrant TH cells exiting the LN to function at distal tissue sites and TFH cells relocating to T-B borders and interfollicular regions. E: Cognate contact between pre-GC TFH cells and antigen-primed B cells is required for multiple programming events in the pathway to B-cell immunity. F: Clonal expansion, antibody class switch, and non-GC plasma cell development proceeds within extrafollicular regions of LNs. G: Secondary follicle formation and antibody class switch precede formation of the GC reaction as the dominant pathway to memory B-cell formation. |
B-cell immunity is initiated on two fronts. Initially, naïve B cells are activated through cell-associated antigen to uptake antigen, process and present peptide major histocompatibility complex [MHC] class II (pMHCII) complexes to enable cognate contact with pMHCII-specific TFH cells. These antigen-primed B cells relocate to T-B-cell borders in lymphoid tissues to increase the likelihood of contact with antigenprimed TFH cells. On the second front, populations of dendritic cells (DCs) also take up antigen, process and present pMHCII, and migrate to draining lymphoid tissues to initiate TFH-cell responses. One outcome of contact with pMHCII-expressing DCs is differentiation of the TFH-cell lineage and the productive contact between pMHCII-specific TFH cells and antigen-primed B cells. This pre-GC cell contact and bidirectional exchange of molecular programming is central to the development of effective B-cell immunity. Antibody class switch, extrafollicular plasma cell differentiation, and initiation of the GC reaction are major B-cell fates associated with this initial phase in B-cell immunity (Fig. 10.1).
Antigen Presentation to B Cells
B cells can acquire soluble antigen by free diffusion into lymphoid follicles1 or through the lymphoid system of conduits.2 However, populations of lymph node (LN) subcapsular sinus (SCS) macrophages appear most effective at presenting cell-associated antigen to follicular B cells.3 B cells take up noncognate antigen presented by SCS macrophages through complement receptors and transfer it into follicular regions and onto follicular DCs (FDCs), which can serve as a source of antigen to prime naïve B cells.4 In contrast, priming with the cognate antigen at first contact with SCS macrophages results in movement of antigen-specific B cells to the T-B-cell borders and antigen-specific B-cell responses to captured antigens.4,5,6 Hence, the SCS macrophages filtering the lymphatic fluid not only protect from systemic infection,7 but also effectively initiate T helper cell-regulated antigen-specific B-cell immunity.
DCs can also effectively prime B-cell immunity. Injection of DCs pulsed with protein antigen can induce isotype switch and promote efficient B-cell responses. B cells can form synapse-like interactions with antigen-pulsed DCs.8,9 More recently, two-photon imaging revealed that naïve B cells entering local LNs surveyed protein antigen-pulsed DCs before entering the follicular areas.10 In this model, engagement of BCR led to calcium flux, migration arrest, and the local accumulation of the antigen-specific B cells. Furthermore, there is a reticular network of collagen fibers that physically connects the subcapsular and paracortical sinuses of LNs to blood vessels and separates these regions from T- and B-cell areas.11 This organization facilitates the efficient delivery of
soluble antigen toward the lumen of high endothelial venules without entering the LN parenchyma. While resident DCs can access this conduit transport system, follicular B cells may have difficulty accessing soluble antigen.
soluble antigen toward the lumen of high endothelial venules without entering the LN parenchyma. While resident DCs can access this conduit transport system, follicular B cells may have difficulty accessing soluble antigen.
Nevertheless, in the context of protein antigens, B cells must recognize their cognate antigen and internalize, process, and present peptides from this antigen in the context of MHCII in order to receive pMHCII-specific T-cell help. If the antigen is cell associated, in the presence of adequate costimulation, some aspects of the B-cell response can proceed in a TI manner. B-cell proliferation and plasma cell development can occur in the absence of cluster of differentiation (CD)40-CD40L interactions with some residual isotype switch induced by the action of TACI and BAFF-R.12 Thus, B cells can respond in a TI manner to protein antigens but do not express the characteristic range of outcomes and the full extent of protective immunity that is found with cognate regulation and T-cell help.
Antigen-Specific B-Cell Activation
Initial activation of naïve B cells through the BCR triggers multiple gene expression programs that enable effective contact with cognate T helper cells. Dynamic contacts with membrane-associated antigens determines the amount of antigen naïve B cells accumulate following antigen exposure.13 Effective cell contacts require expression of the signaling adaptor dedicator of cytokinesis 8 (DOCK8).14 Mutations in DOCK8 disrupted integrin ligand accumulation in the immune synapse without altering BCR signaling events. B-cell-specific conditional ablation of the calcineurin regulatory subunit 1 (CNB1),15 myocyte enhancer factor 2c,16,17 and stromal interaction molecule 1 and stromal interaction molecule 218 have shown that calcium responsiveness is necessary for cell cycle progression in these early pre-GC stages of B-cell responses. Hydrogen voltage-gated proton channels 1, which are internalized with the BCR, have been recently implicated in early B-cell programming events.19 Single-pulsed BCR signaling20 that only partially activated NF-κB increased CC chemokine receptor 7 and MHC class II expression, and responsiveness to CD40, indicating some of the early facets of B-cell activation. Severe defects in early B-cell proliferation have also implicated integrin-binding CD98hc21 and extracellular signal-regulated kinase activation22 in preparing the antigen-primed B cells to receive cognate T-cell help in vivo. Hence, initial antigen recognition, uptake, processing, and presentation critically impact the early B-cell developmental fate. High-resolution dynamic imaging has provided substantial insight into the earliest events associated with initial BCR engagement on naïve B cells. The membrane cytoskeleton controls BCR diffusion; disruption of this organization initiates signaling.23,24 Discrete microclusters form upon antigen binding25 and recruit multiple components of the intracellular BCR signaling network to initiate signal transduction.26 One rapid response involves B-cell spreading to increase surface contact with antigen on the presenting cells.13 Central clustered antigen at the cellular interface is then internalized into antigen-processing lysosomes for presentation with MHCII8 as the cognate point of TFH contact. Curiously, antigen presentation appears asymmetrically segregated with one daughter retaining larger antigen stores thereby more able to contact pMHCII-specific TFH cells.27 Whether these developmental outcomes are directed by initial context of antigen presentation or stochastically assorted28 remains an interesting fundamental issue with an early impact on antigen-specific B-cell development.
Dendritic Cell Maturation
DCs are essential antigen-presenting cells (APCs) for initiating adaptive immunity. Multiple DC subsets exist prior to antigen challenge. Different phenotypic schemes can be used to characterize DC subsets with the origins and developmental relatedness of different DC subsets still subject to debate.29,30 In the murine system, three main bone marrow-derived DC subsets enter all secondary lymphoid organs via the blood and reside at different levels. These bloodderived DCs all express CD11c and are distinguishable as CD11bhi CD8aneg DCs, CD8ahi DCs, and 6B2hi plasmacytoid DCs. In LNs draining the skin, there are at least two further CD11c+ DC subsets, Langerhans cells (LCs) and dermal DCs, that emigrate from the skin, even at homeostasis.31 Thus, before antigen challenge, multiple subsets of DCs are available to differentially process and present antigen to the adaptive immune compartment.
Protein antigen administration in the absence of inflammation induces immune tolerance. In contrast, coadministration of an immune adjuvant activates facets of innate immunity, induces inflammation, and primes antigenspecific adaptive immunity. Sensing pathogens involves pattern recognition receptors such as the evolutionarily conserved toll-like receptors32,33 and the more recently described nucleotide oligomerization domain-like receptors.34 Most forms of antigen and innate stimulators require DC priming at some level for B-cell immunity, whereas particulate antigen in virus-like particles are more reliant on B-cell innate receptor activation.35 Nevertheless, both families of innate receptors recognize different types of microbial components initiating programs of DC maturation that promote immediate local inflammation and innate effector clearance mechanisms.
Temporal and spatial constraints on DC maturation provides another layer of regulation for the innate system that can impact adaptive immunity. In the steady state, DCs form dense networks at the T-B borders of LNs. Interestingly, motile lipopolysaccharide-activated DC immigrants will rapidly coalesce with this preexisting network in vivo.36 In separate studies using genetically tagged LCs, the emigrants of LCs and dermal DCs were shown to emerge separately in time and colonize separate regions of the T-B border.37 A similar temporal regulation was seen using antibodies to specific pMHCII complexes with resident DCs presenting an early wave of pMHCII and dermal DCs emerging later in a second wave.38 In each of these studies, the immune stimulus was varied and the coordinated response of the innate system also qualitatively and quantitatively different.
Clonal Selection in Helper T Cells
The initial outcome for pMHCII+ DC interactions with naive TH cells is T-cell receptor (TCR)-driven clonal selection. TH– cell responses that focus the specific TH-cell response to a set of dominant TCRs provide access to the mechanisms that underpin TH clonal selection.39,40,41 Earlier studies indicate the importance of TCR-pMHCII affinity in determining TH cell fate.39,42,43,44 In this model, selective expansion of clonotypes expressing higher-affinity TCRs would rely on competition for pMHCII complexes on APCs. Alternatively, in vitro studies using altered peptide ligands suggested that the duration of TCR-pMHCII contact was critical to cell fate and therefore defined “best fit” in vivo.45,46,47,48 In support of this model, TH cells expressing TCR with fast off rates were lost over time in vivo.49 Further, there is evidence for different peptides stabilizing the pMHCII to create a hierarchy of dominant peptides.50 Each and all of these variables would impact what is considered “best fit” and influence the outcome of clonal selection. More recently, we provide evidence for a model of TH clonal selection that is based on a TCR affinity threshold.51 Multiple affinity-based thresholds underpin cell fate and TH clonal expansion52 with evidence that the highest affinity TCR assort into the TFH compartment.53 Hence, selection thresholds generate antigen-specific clonal diversity in ways that impact the development of effector TH cell function in vivo.
Follicular Helper T Cells
There were early reports of effector TH cells specialized to regulate B-cell responsiveness. CXCR5 expression was first reported on CD4+CD45RO+ cells in the peripheral blood and secondary lymphoid tissue in humans.54 Gene ablation studies emphasized the role of CXCR5 and CC chemokine receptor 7 in the correct positioning of T and B cells in secondary lymphoid tissue that was also needed to support effective TH cell-dependent B-cell immunity.55,56 Blocking CD28 and OX40 interactions in vivo blocked the development of CXCR5+ TH cells and the GC reaction.57 Further, CXCR5 expression was induced in an antigen-specific manner on TH cells in vivo, and these cells relocated to follicular areas and the GC of responding lymphoid tissue.58 CXCR5+ TH cells were sorted from human tonsil and shown to support antibody production in vitro.59,60 The tonsillar CXCR5+ TH cells expressed high levels of CD40L and ICOS and were found in both the follicular mantle and GC. Adoptive transfers distinguished CXCR5+ B cell helper activity (TFH) from P-selectin ligandhi tissue homing inflammatory mediation (DTH-promoting TH) that emerged together from the same set of precursors in vivo.61 The term TFH cells was coined to categorize this functionally and phenotypically distinct effector TH cell compartment.59,60
As the name implies, the cardinal characteristic of all TFH cells is their repositioning into the follicular regions of secondary lymphoid tissues. Early assessments of cytokine production by in vitro restimulated CXCR5+ TFH cells indicated interleukin (IL)-2, interferon (IFN)-γ, and IL-10 from human peripheral blood60 with evidence for IL-4 and IFN-γ from TCR transgenic mouse TFH cells.61 Early microarray analyses suggested separable gene expression programs for TFH cells and other known TH-cell subsets. CXCL13 was highlighted early62 with evidence for ICOS, IL-21,63,64 IL-21R,65 and the differential expression of Bcl-663 being used as the most reliable attributes of TFH function in vivo. Thus, acquisition of special pre-GC TFH-cell functions was associated with the programming of a separate TH-cell lineage.
Molecules important in the development of normal B-cell immunity were implicated in early studies of TFH development. CD28 deficiency or treatment with blocking CD28 antibodies led to profound defects in B-cell immunity.66 CD28 was required early to initiate naive TH-cell responsiveness to pMHCII+ CD80- and CD86-expressing DCs. In contrast, CD40-CD40L interactions were central to the delivery of T-cell help to B cells.67,68,69 The CD28 family member ICOS70 was implicated in TFH-cell function in pre-GC interactions with pMHCII-expressing B cells.71,72,73 ICOS-deficient humans and mice74,75,76 and ICOS-L-deficient mice had marked deficits in all aspects of B-cell immunity. ICOS deficiency was associated with decreased TFH-cell development and considered an important molecule in the delivery of effector TFH function.77,78 Conversely, overexpression of ICOS in mice with a regulatory defect in ICOS expression79 resulted in an overproduction of CXCR5+ TFH cells and breakthrough autoimmune disease.64 Recent studies indicated that ICOS can substitute for CD28 and rescue the TFH defects and B-cell defects in CD28-deficient mice.80 Furthermore, the abundant CXCR5+ TFH cells in this model act in a T-cell autonomous manner to promote autoantibody production.81 PD-1, another CD28 family member that has been implicated in the negative regulation of chronically activated T cells, was also found on GC TFH cells in human tonsil and mouse.82 Positive and negative influences of antigen-specific TFH-cell costimulation are balanced in ways that remain poorly understood in vivo and are an active avenue of current research in this field. BCL6 is required for development of the TFH program83,84,85 and is reinforced within TFH cells upon pre-GC B-cell contact.86,87 IL-21 also plays a major role in TFH function with substantial loss of B-cell immunity in its absence.88,89 Recent studies have identified BCL6 in antigen-specific TFH cells and BLIMP1, which has an opposing function, in non-TFH cells.53 BCL6 and BLIMP1 expression was mutually exclusive across these two TH-cell subtypes, already evident by the second cell division in vivo.29 More recently, transcription factors c-Maf and BATF were shown to act with BCL6 to program TFH development.90,91 Hence, distinct transcriptional programming of unique cellular functions directs the early TFH-cell development that is central to subsequent B-cell immunity (Fig. 10.2).
Pre-Germinal Center Follicular Helper T-B-Cell Contact
First contact between antigen-specific TFH and antigenprimed B cells has also been captured through dynamic imaging.92 Stable “monogamous” interactions between one antigen-specific TFH cell and one B cell can last for a duration of 10 to 60 minutes in the follicular regions of the LNs. These interactions were accompanied by highly dynamic movements, with the B cells migrating extensively and leading
the TFH cells.92 A recent study demonstrated that expression of the adaptor molecule signaling lymphocytic activation molecule-associated protein (SAP) was needed to form long duration contacts with antigen-primed B cells.93 In these studies, there was no role for SAP in early TFH-DC contacts, and the SAP-deficient TFH cells reached the follicular regions and expressed all the hallmarks of pre-GC TFH cells (CXCR5hi, CD40L+, ICOShi, and OX40hi).93 Furthermore, in the absence of SAP, TFH cells were still capable of cytokine production94 but unable to promote GC formation.95,96 More recently, dynamic imaging places ongoing critical pre-GC contacts in the interfollicular zones of LNs97 with a requirement for persistent BCL6 expression in B cells to maintain effective cognate contact.98 Therefore, early TFH-cell developmental programs establish the capacity for cognate contact needed to promote ongoing antigen-specific B-cell immunity.
the TFH cells.92 A recent study demonstrated that expression of the adaptor molecule signaling lymphocytic activation molecule-associated protein (SAP) was needed to form long duration contacts with antigen-primed B cells.93 In these studies, there was no role for SAP in early TFH-DC contacts, and the SAP-deficient TFH cells reached the follicular regions and expressed all the hallmarks of pre-GC TFH cells (CXCR5hi, CD40L+, ICOShi, and OX40hi).93 Furthermore, in the absence of SAP, TFH cells were still capable of cytokine production94 but unable to promote GC formation.95,96 More recently, dynamic imaging places ongoing critical pre-GC contacts in the interfollicular zones of LNs97 with a requirement for persistent BCL6 expression in B cells to maintain effective cognate contact.98 Therefore, early TFH-cell developmental programs establish the capacity for cognate contact needed to promote ongoing antigen-specific B-cell immunity.
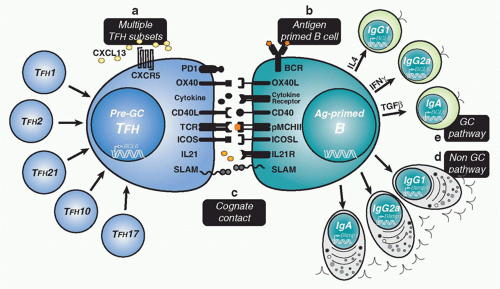 FIG. 10.2. Pre-Germinal Center (GC) Follicular Helper T (TFH)-B-Cell Contact. A: Multiple subsets of antigen-specific pre-GC TFH are produced to regulate B-cell immunity. The organization of function within TFH subsets remains speculative. There is evidence for different cytokine-secreting TFH cells that regulate commitment to separate antibody class as well as different types of TFH cells that regulate non-GC plasma cell differentiation. Bcl-6 and CXCR5 expression is thought to be a common feature of all TFH cell subsets. B: Antigen-primed B cells must process and present peptide major histocompatibility class II to receive cognate help from pre-GC TFH cells. Upregulation of molecules involved in helper T cell contact is a poorly resolved component of early antigen-driven B-cell maturation. C: Cognate contact between antigen-specific T-cell receptor and peptide major histocompatibility class II complexes focus pre-GC TFH-B cell intercellular exchange of molecular information. Modifying interactions at first contact are known to involve costimulatory molecules (eg, cluster of differentiation [CD]40L-CD40, ICOS-ICOSL), accessory interactions (eg, signaling lymphocytic activation molecule family interactions, OX40-OX40L), cytokine and cytokine receptors (eg, IL4-IL4R, interferon [IFN]γ-IFNγ receptor, IL21-IL21R). Distribution of these functional attributes within pre-GC TFH compartments is not yet well resolved in vivo. D: The non-GC pathway to plasma cell development permits antibody class switch recombination (CSR) without somatic hypermutation (SHM) depending largely on the cytokine stimulus provide by pre-GC TFH cells. Blimp-1 expression is required for plasma cell commitment across all antibody classes. E: The GC pathway to memory B-cell development begins with extensive B-cell proliferation within secondary follicles that will polarize into dark and light zone to initiate the GC reaction. The GC pathway is associated with Bcl-6 upregulation and activationinduced cytidine deaminase expression to supports both antibody CSR and SHM. These GC features emerge across all antibody classes and require productive and long duration pre-GC TFH contact. |
It has been unclear how differential BCR affinity can impact the early fate of antigen-primed B cells. Very-low-affinity B cells are capable of forming GCs99 but fail to do so in the presence of high-affinity competition.100 In contrast, there is evidence for the highest-affinity B cells preferentially entering the non-GC plasma cell pathway, leaving lower-affinity B cells to mature within the GC cycle.101 This issue has been addressed more recently using intravital imaging to examine the early pre-GC selection events.102 In this model, access to antigen was not impacted by BCR affinity, but the differential capacity to present antigen to pre-GC TFH cells assorted with BCR affinity. Increased T-cell help promoted greater access to both the plasma cell pathway and the GC reaction. Thus, BCR affinity thresholds regulate B-cell fate at the earliest pre-GC junctures of antigen-specific TFH-B interactions. Overall, the precise function of different costimulatory molecules and their combinatorial impact on antigen-specific B cell fate remains an exciting area of current interest. Quantitative differences in cell surface molecules in combination with mixtures of cytokines will likely synergize in
predetermined ways to skew pMHCII-expressing B cells into separate pathways of B-cell immunity. Unraveling these molecular combinations will help to define the rules of molecular control for antigen-specific B-cell immunity.
predetermined ways to skew pMHCII-expressing B cells into separate pathways of B-cell immunity. Unraveling these molecular combinations will help to define the rules of molecular control for antigen-specific B-cell immunity.
Controlling Antibody Class
Pre-GC cognate TFH-cell contact controls multiple antigenspecific B-cell differentiation options. Beyond this early pre-GC developmental juncture, antibody class switch appears in both in the extrafollicular pathway to plasma cell differentiation and the GC pathway to memory development.103 Class switch also requires multiple rounds of cell division in the target cell before expression of non-immunoglobulin (Ig)M antibody.104 Hence, it is likely that initial commitment to antibody class occurs at the first pre-GC point of cognate contact and is propagated in a lineal manner across each developmental option (see Fig. 10.2).
Class switch recombination (CSR) is an intrachromosonal deletional process between the switch (S) regions that reside 5′ of each constant region gene in B cells (except Cδ).105 Signaling through CD40 and cytokine receptors induces germline transcription through the targeted S regions providing activation-induced cytidine deaminase (AID; also known as Aicda) access to deaminate cytosines in the single stranded template. AID is required and sufficient for the initiation of the CSR reaction in the activated locus.106 AID-deficient animals107 and humans108 display no CSR or somatic hypermutation (SHM) of the Ig genes. Recent evidence indicates that following antigen stimulation, AID expression is regulated in B cells by paired box gene/protein 5, E-box proteins,109 homeobox C4,110 and forkhead box O1.111 Removal of the resulting uracils and deoxyribonucleic acid (DNA) cleavage generates double strand breaks that trigger the recruitment of DNA damage machinery, mismatch repair, and nonhomologous end joining to complete the CSR event.112 The adapter protein 14.3.3 is recruited with AID to switch regions113 and polymerase ζ has been implicated in the repair process associated with CSR.114 Peripheral B cells undergoing CSR in the absence of the x-ray-repair crosscomplementing protein 4 component of the double strand break repair machinery are also highly susceptible to translocation events and oncogenic transformation.115 Antibody class switch is a destabilizing and potentially dangerous cellular event that can proceed without SHM in the extrafollicular pathway.
Induced CD40L on pre-GC TFH cells and the receipt of this signal through CD40 on B cells is required for antibody class switch.67,116 Animals and humans lacking CD40 display a hyper-IgM syndrome with profound defects in class switch, GC formation, and the development of affinity matured B-cell memory.116 ICOS expression on activated TFH cells is thought to act upstream of CD40L in this temporally orchestrated set of events.117 ICOS-deficient animals also have clear defects in antibody class switch, GC formation, and the development of B-cell memory.74,75,76 Some residual class switch in the absence of CD40/CD40L interactions may be explained by the action of TACI and BAFF-R.12 OX40/OX40L interactions also quantitatively impact class switch while CD27-CD70 interactions promote plasma cell (PC) production.118 Thus, the range of molecules expressed at the pre-GC TFH cell surface influences the developmental impact of TCR-pMHCII contact on antigen-primed B cells. Considering the range of antibodies that can be produced, there must be multiple subtypes of pre-GC TFH cells that control antibody class. Different classes of pre-GC TFH would vary in production of TFH cell-derived cytokines to control antibody isotype. IL-4 and IFN-γ are reciprocal regulators of IgG1 and IgG2a production.119 Animals lacking IL-4 or Stat 6 have decreased IgG1 levels and no IgE.120,121 IL-4 also acts together with IL-21 to control IgG subtypes and IgE levels.88 In contrast, transforming growth factor (TGF)β is implicated in the induction IgA, while IL-2 and IL-5 augment IgA production.122 Similarly, IL-6 may selectively support IgG2a and IgG2b expressing B cells in vivo.123 Each of these factors can exert their effects in vitro or in a bystander manner in vivo. However, it is thought that the directed delivery of these soluble molecules toward points of TCR-pMHCII contact allows soluble signals to focus locally in an antigen-specific cognate manner.
Within antigen-responsive B cells, the molecular machinery that regulates CSR is deployed in an antibody class-specific manner. The global CSR machinery is targeted by transcription factors downstream of the cytokine receptors that control specific antibody classes. For example, IFNγ activates signal transducer and activator of transcription downstream of the IFNγ receptor to induce T-bet and promote IgG2a class switching.124,125 Similarly, TGFβ signals through TGFβ-receptor to activate SMAD and RUNX transcription factors to promote IgA class switch.126 Furthermore, the transcriptional regulator BATF required for TFH development is also required in B cells to generate germline switch transcripts and promote AID expression.91 Finally, Ikaros regulates antibody class decisions by differentially controlling transcriptional accessibility of constant region genes.127 How the initial commitment to antibody class is maintained and propagated during clonal expansion, BCR diversification, and affinity-based selection within the GC reaction remains an important but unresolved issue. Therefore, it remains plausible that functional reprogramming accompanies CSR creating separable lineages of class-specific memory B cells in vivo.
Extrafollicular Plasma Cell Development
Under the cognate regulation of pre-GC TFH cells, a cohort of antigen-primed B cells clonally expand within the T-cell zones of secondary lymphoid organs and rapidly give rise to PCs. Within the first few days after antigen exposure, small foci of B-cell blasts can be seen within the T-cell zones.103 This plasmablast stage appears transitional and defines pre-PCs that may secrete antibody but also retain the capacity to proliferate. In contrast, PCs are typically considered terminally differentiated and in a postmitotic state.128,129 PCs display a marked increase in IgH and IgL messenger ribonucleic acid and prominent amounts of rough endoplasmic reticulum to accommodate translation and secretion of abundant Ig. They have reduced or lost numerous cell surface molecules
including B220, CD19, CD21, and CD22 with an increase in the proteoglycan syndecan-1 (CD138),130,131 often used as a distinguishing marker for PCs. We recently demonstrated that class-switched plasma cells retained robust antigenpresentation capacity as a negative feedback loop for regulation of TFH cells.132 These studies further highlight the bidirectional programming between antigen-primed B cells and pMHCII-specific TFH cells during developing immune responses.
including B220, CD19, CD21, and CD22 with an increase in the proteoglycan syndecan-1 (CD138),130,131 often used as a distinguishing marker for PCs. We recently demonstrated that class-switched plasma cells retained robust antigenpresentation capacity as a negative feedback loop for regulation of TFH cells.132 These studies further highlight the bidirectional programming between antigen-primed B cells and pMHCII-specific TFH cells during developing immune responses.
In mice deficient for SAP, the extrafollicular pathway to PC development, remains largely intact but the GC pathway is blocked.95 Recent studies indicate that SAP-deficient TFH cells exhibit heightened CD40L expression but decreased ICOS induction that alone can account for this defect.94 More recent studies indicate that SAP-deficient TFH cells can only form short duration cognate contact with antigen-primed B cells, which are insufficient for GC entry but adequate for PC formation. It will be important to dissect the separable signals and cellular subsets of pre-GC TFH cells that regulate the rapid but short-lived effector B-cell response in vivo. Extrafollicular PCs have half-lives of 3 to 5 days133 and express germline-encoded antigen-specific antibodies.131,134 Newly formed PCs migrate via the marginal zone bridging channels into the red pulp of the spleen and into the medullary cords of LNs. Migration patterns in vivo appear controlled by increased responsiveness to the CXCR4 ligand CXCL12 and decreased expression of CXCR5 and CC chemokine receptor 7.135 There is evidence for BCR affinity-based selection even at this early stage in the response.131 In some studies, B cells expressing low affinity BCR remain non-GC and high-affinity B cells preferentially enter the GC pathway. 100 The converse can also be seen with high-affinity B cells preferentially entering the non-GC short-lived PC pathway.101 While seemingly contradictory, these different outcomes in vivo may reflect the plasticity of intercellular control at the TFH-B point of contact.
The transcriptional control of PC development also exists in multiple layers controlling a cascade of developmental change. The transcriptional repressor prdm-1 (encoding Blimp-1) plays a central role in the regulation of PC development.136 B cells lacking Blimp-1 do not differentiate into extrafollicular PC or post-GC PC, with TI and TD antibody responses profoundly diminished in vivo.137 These Blimp-1-deficient animals also display defective levels of serum antibody, suggesting that spontaneous production of antibody by B-1-B cells also requires Blimp-1 expression. Structurefunction analysis indicates the modular action of Blimp-1 integrating a variety of environmental cues that lead to PC development.138,139 Blimp-1 represses proliferation through c-myc as one direct target among many others involved in cell cycle control. Blimp-1 also induces antibody secretion by repressing the transcription factor Pax-5, thereby derepressing Xbp-1. The transcription factor Xbp-1 controls the unfolded protein response, and many facets of the cellular secretory mechanism that are critical to PC function and survival.140,141,142
The transcriptional coactivator OBF-1 also appears important for B cells to complete the PC program.143 In the absence of OBF-1, Bcl-6, Pax-5, and AID are not repressed, blocking the induction of Blimp-1 and PC development. In contrast, ablation of the transcription factor MitF leads to the spontaneous development of PC that appears independent of antigen stimuli.144 Interestingly, reexpression of bcl-6 and its cofactor MTA3 re-activates the B-cell program and increased CD19 and MHC II, and decreased CD138 in plasma cell lines.145 This remarkable study suggests that PC fate is not as terminal and passive as it has been thought to be, and that the PC fate remains subject to dynamic gene expression programs. Regulation of the unfolded-protein response by X-box-binding protein 1 is not needed for plasma cell development but is necessary for antibody secretion.146,147 Epstein-Barr virus-induced molecule 2 also appears essential for B-cell movement to extrafollicular sites and the non-GC plasma cell response.148,149 In addition, Epstein-Barr virus-induced molecule 2 guides recently activated B cells to interfollicular LN regions and then to outer follicular areas as a prelude to GC formation. Furthermore, there appears to be an early pre-GC proliferative phase at the perimeter of follicles that also precedes GC formation and BCR diversification.150 Interestingly, recent dynamic imaging studies indicate migration of TFH cells to the follicle interior, even before accumulation of GC B cells.97
THE GERMINAL CENTER CYCLE
Movement of antigen-primed B cells into the follicular regions of lymphoid organs after effector TFH-cell contact initiates the GC pathway to memory B-cell development. These pre-GC B cells expand rapidly within the follicular region to form areas of B220+IgDlow antigen-specific B cells referred to as secondary follicles. Secondary follicles polarize into T-cell proximal dark zones of cycling centroblasts and opposing light zones of largely noncycling centrocytes among the dense processes of FDC and sparse presence of GC TFH cells.151,152 This broad anatomical distribution of the GC reaction orients the activities of clonal expansion, BCR diversification, and clonal variant selection that underpin the evolution of high-affinity memory B cells and post-GC plasma cells in this pathway (Fig. 10.3). Under normal physiologic conditions, the GC reaction emerges as the most efficient means to control affinity maturation and memory B-cell development. However, in the disorganized or absent preimmune lymphoid subcompartments of mice lacking LTa, LTb, tumor necrosis factor receptor I, and LTbR, the activities of the GC reaction remain disorganized but largely intact.153,154,155,156 Vestiges of the GC or small cell aggregates manage the expansion, diversification, and selection steps required for affinity maturation and memory B-cell development.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


