that the activation of B cells by antigen is far more dynamic than originally thought. It has been possible to infer from the behavior of the B cells and BCRs in these images the mechanisms underlying antigen-driven events that lead to B-cell activation. Such images are changing our view of BCR-mediated B-cell activation and are providing the tools to gain an understanding of the mechanisms underlying diseases that result from inappropriate B-cell activation including systemic autoimmune diseases and certain B-cell tumors.
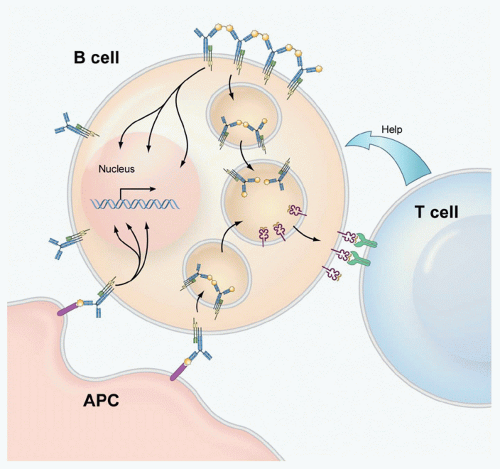 FIG. 9.1. Overview of the Activation of B Cells by Antigen. B-cell activation is initiated by the binding of antigen to the B-cell receptor (BCR). B cells encounter antigen either in solution or on the surface of an antigen-presenting cell (APC), likely as complement coupled antigens or as immune complexes bound to APC complement receptors or FcRs. Antigen binding triggers signaling cascades that lead to the activation of a variety of genes associated with B-cell activation. The BCR is also a trafficking receptor; under the influence of BCR signals, it transports antigens into intracellular compartments where the antigens are processed and presented on major histocompatibility complex class II molecules for recognition by helper T cells. |
Concerning the impact of the environmental context in which B cells are activated, this chapter will focus on the effect of B-cell coreceptors that directly affect BCR signaling. We will provide a comprehensive list of the coreceptors and what they respond to and describe how two of the best studied coreceptors, the enhancing cluster of differentiation (CD)19/CD21 complex and the inhibitory FcγRIIB receptor, function to influence B-cell responses. We will also comment on the interactions between the innate immune system’s toll-like receptors (TLRs) and the BCR that appear to serve to regulate each other’s signaling.
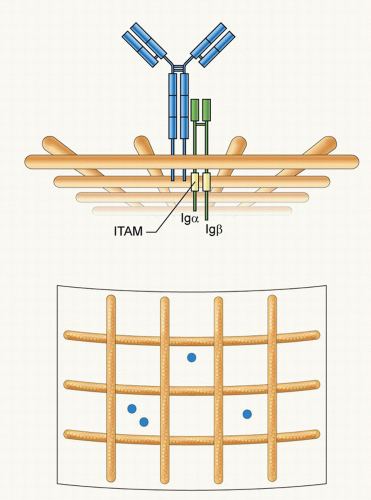 FIG. 9.2. The Structure and Organization of the B-Cell Receptor (BCR). The immunoglobulin (Ig)M BCR is composed of a membrane form of IgM associated with a covalently linked heterodimer of Igα and Igβ that contain in their cytoplasmic domains immunoreceptor tyrosine-based activation motifs. The BCR is depicted compartmentalized on the plasma membrane by actin cytoskeleton “fence”9 as proposed by Batista et al.21 Two different views are provided: a side view showing the BCR short cytoplasmic tail and the Igα-Igβ tails in the cytosol and a view from above showing the BCRs compartmentalized by actin “fences.” |
dynamic structures that can disassemble and reassemble during B-cell activation.
These BCR microclusters are enriched in tyrosine phosphorylated proteins and are the site of Lyn and Syk recruitment and thus appear to be the elemental signaling units. B cells subsequently spread over the bilayer allowing the engagement of additional antigens (see Fig. 9.3). Following maximal spreading, the BCR-antigen microclusters are actively moved toward the center of the contact area, and the B cell contracts to form an immunologic synapse. This dynamic process of touching, spreading, and contraction occurs within minutes of the BCR’s first contact with the bilayer.23 These observations bring us back to the fundamental questions: How are BCR microclusters formed, and how do BCR microclusters trigger BCR signaling? Our understanding of these processes is still incomplete. However, based on current data, several models have been proposed that address these questions.
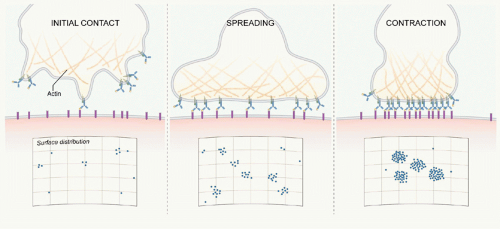 FIG. 9.3. The Activation of B Cells on Antigen-Presenting Cell (APC) Surfaces. The initial contact of the B cell with an antigen-containing APC surface is through finger-like protrusions of the membrane. Antigen binding induces B-cell receptor (BCR) clustering and signaling that triggers the B cell to spread over the APC surface, forming additional BCR clusters as antigens are encountered. After maximal spreading, the B cell contracts, actively moving BCR clusters to the center of the contact area ultimately forming an immune synapse. |
adaptors in B-cell activation, leading to important insights into how they integrate BCR signaling. Although adaptors generally lack any enzymatic activity, they consist of multiple protein-protein or protein-lipid interaction domains, including SH2, SH3, PH and PX homology domains (see Box 9.1). Each of these domains has the potential to interact with a number of proteins that are critical to amplify BCR signaling by facilitating the coupling of multiple downstream signaling pathways. Essentially, the adaptors define where and when macromolecular complexes are assembled, allowing both spatial and temporal regulation of signaling cascades. To illustrate the importance of adaptors, we describe the functions of three: B-cell linker protein (BLNK); B-cell adaptor for PI3K (BCAP), and B-cell adaptor molecule of 32 kDa (Bam32).
TABLE 9.1 The Phenotype of Mice Deficient in Key B-Cell Signaling Components | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
calcium responses. Although Syk can directly phosphorylate PLC-γ2 in vitro,57 expression of a functional BCR, Lyn, and Syk in nonlymphoid cells did not induce PLC-γ2 phosphorylation or calcium mobilization,58 leading to the discovery of the B-cell-specific adaptor protein, BLNK. BLNK (also known as SLP-65 or BASH) is a cytoplasmic protein consisting of an N-terminal region containing a short leucine zipper motif, a proline-rich region within the middle third of the molecule, and a C-terminal SH2 domain. BLNK contains 13 tyrosine residues, of which 6 are in putative SH2 binding motifs and are phosphorylated upon BCR clustering. BLNK is recruited to the clustered BCR by binding through its SH2 domain to the phosphorylated non-ITAM tyrosine 204 in Igα. Mice with a Igα Y204 mutation exhibit reduced BLNK phosphorylation and calcium fluxes.59 Once translocated to the plasma membrane, BLNK is phosphorylated by Syk, which creates docking sites for SH2 domains of multiple effector molecules facilitating interactions among them and allowing them to phosphorylate and activate their respective signaling pathways. One of the best studied examples of how BLNK bridges multiple signaling components is provided by the activation of PLC-γ2. PLC-γ2 activation is completely abolished in Syk-deficient B cells.60 In addition, BCR-induced PLC-γ2 activation is diminished in Btk-deficient B cells, suggesting that both Syk and Btk are required for PLC-γ2 activation.61 However, the mechanisms underlying how these two families of PTKs regulate PLC-γ2 activation became clear only after the discovery of BLNK. After phosphorylation by Syk, BLNK creates binding sites for the SH2 domains of both Btk and PLC-γ2, bringing them into close proximity with each other, and hence facilitating PLC-γ2 phosphorylation at Tyr753 and Tyr759 by Btk, which is required for PLC-γ2 activation.62,63,64,65,66,67,68 Cells expressing a mutant BLNK lacking either Btk or PLC-γ2 binding sites
show significant reduction in calcium activation, further suggesting that BLNK acts as a scaffold for bridging Btk to PLC-γ2.69 A single BLNK molecule can bind three PLC-γ2 molecules, thus BLNK also functions as an amplifier of BCR signaling.62 Following BCR clustering, both BLNK and Btk are also translocated to the plasma membrane bringing along PLC-γ2 and allowing it to gain access to its substrate PI(4,5)P2 in the inner leaflet of the plasma membrane.66,67,68 In addition to phosphorylating PLC-γ2 and recruiting it to the plasma membrane, Btk also recruits PIP5 kinase to the plasma membrane, which catalyzes PI(4,5)P2 synthesis from PIP(4)P to ensure that activated PLC-γ2 does not run out of its substrate.70 Once activated, PLC-γ2 hydrolyzes PI(4,5) P2 to generate two important second messengers: inositol trisphosphate (IP3) and diacyl glycerol (DAG). BLNK not only couples Btk and PLC-γ2, but it also binds to the SH2 domains of Vav and Nck, resulting in activating the mitogen-activated protein (MAP) kinase pathway, cytoskeletal rearrangements, and BCR internalization.71
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


