development, as shown in Figure 8.2. Early precursors can also be found in the fetal omentum.9 In contrast with the bone marrow, cells at most differentiation stages in the fetal liver appear to be rapidly proliferating so that larger and larger numbers of B-lineage cells are detected at progressive days of gestation. Another distinction of fetal liver from bone marrow development in the adult is the absence of terminal deoxynucleotidyl transferase (TdT),10,11 an enzyme that mediates nontemplated addition of nucleotides at the D-J and V-D junctions of Ig heavy chain. 12,13,14 Therefore, heavy chains produced during fetal development have little or no N-region addition, and CDR3 diversity is constrained even further by favoring of short stretches of homology at the V-D and D-J junctions.15,16 Rearrangement of certain V or D elements may also differ between fetal and adult development, as for example, the reported high utilization of the DFL16.1 segment in fetal liver.17 Differential expression of genes other than TdT also distinguish B-cell development during fetal life from that in the adult including precursor lymphocyte regulated myosin light chain like PLRLC transcripts11,18 and major histocompatibility complex (MHC) class II.19,20 Interestingly, although absence of the cytokine IL-7 completely eliminates bone marrow B-lineage development,21 it nevertheless spares some fetal development,22 suggesting a difference in growth requirements. The B-cell progeny of this early fetal wave may largely consist of B cells quite distinct from adult-derived cells, populating the B-1 subset.23
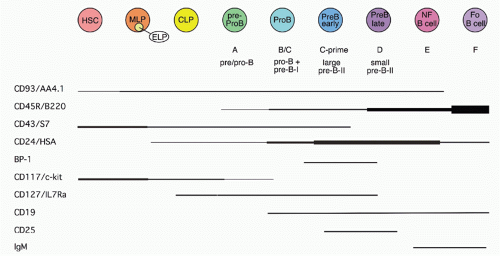 FIG. 8.1. Differentiation Diagram for Development of B Cells from Hematopoietic Stem Cells. Expression of the each surface protein is indicated by a line. Changes in level of expression is indicated by line thickness. CLP, common lymphoid progenitor; ELP, early lymphoid progenitor; Fo B, follicular B cell; HSC, hematopoietic stem cell; MLP, multilineage progenitor; NF B, newly formed B cell. |
progenitors (CLPs).31,32,33 The CLP cell fraction was identified by lack of a panel of “lineage markers,” expression of the IL-7Rα chain, and distinctive, intermediate levels of c-KIT, compared to higher levels on HSCs. Initial characterization of these cells in various functional assays suggested that these cells could generate B, T, natural killer (NK), and a subset of dendritic cells but no other blood cell lineages. The reason for this restriction has been intensively studied, and downregulation of the receptor for granulocyte-myeloid colony stimulating factor has been suggested to be a key event in this process.34 Cells with the phenotype of CLP constitute about 1/3000 of bone marrow cells. Prior to the CLP stage, multipotent progenitors exhibit low-level expression of genes characteristic of diverse cell lineages, leading to the idea that such promiscuous expression indicates chromatin accessibility that facilitates flexibility in cell fate decisions.35
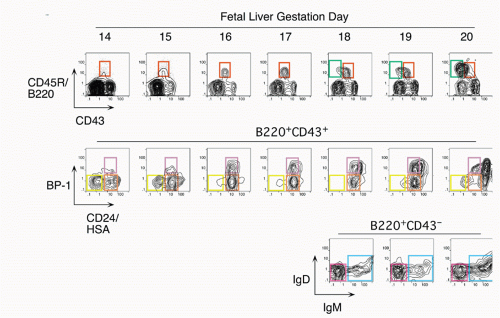 FIG. 8.2. Phenotypic Progression of Developing B-Lineage Cells in Mouse Fetal Liver Analyzed at Different Days of Gestation. Note that B220+cluster of differentiation (CD)43+ cells precede B220+CD43- cells and that within the B220+CD43+ fraction, heat stable antigen (HSA)- cells precede HSA+ cells, and BP-1- cells precede BP-1+ cells. Within the B220+CD43- fraction, the immunoglobulin M+ percentage increases until birth (at about day 20). |
inhibits RUNX function, have revealed that its expression negatively impacts pre-pro-B-cell through pre-B-cell populations in bone marrow.59 Although the frequency of CLP stage cells was unaffected, the expression of B-lineage associated genes (such as CD79a and λ5) was decreased, demonstrating the key importance of early RUNX activity.
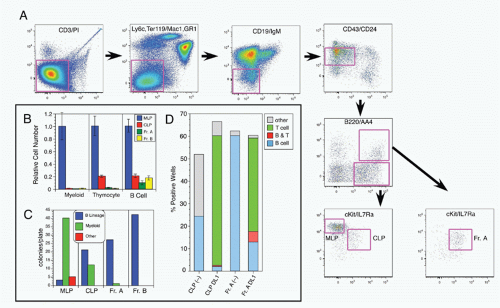 FIG. 8.3. A: An approach for purifying the earliest stage of B-lineage cells in mouse bone marrow. Bone marrow cells expressing cell surface proteins characteristic of differentiated stages of T, myeloid, erythroid, and B lineages are depleted sequentially by electronic gating in the first three panels. Cells with low-level expression of cluster of differentiation (CD)24/heat stable antigen (HSA) and intermediate levels of CD43(S7) are selected in the fourth and the distribution of CD45R/B220 versus CD93/AA4 is shown in the fifth. AA4+B220− cells contain multilineage progenitors (MLPs) and common lymphoid progenitors (CLPs), resolved by analysis for c-Kit versus interleukin (IL)-7Rα in panel six. AA4+B220+ cells, shown in the final panel, are enriched for c-Kit+IL-7Rα+ cells, termed Fr. A. CLP stage cells resemble Fr. A, but lack detectable expression of CD45R/B220. In contrast with CLP and Fr. A, MLP stage cells have higher levels of c-Kit and lack IL-7Rα expression. B: Functional analysis of early B-lineage cells by in vivo competition assay, showing absence of myeloid or T-lineage generation, but production of B-lineage cells from Fr.A. In contrast, CLP stage cells generate B and T cells, whereas MLPs repopulate B, T, and myeloid lineage cells. MLP, CLP, and Fr. A as identified in A. Fr. B stage cells are DJ/DJ rearranged pro-B cells, identified as CD19+CD43+CD24(HSA)+. C: Functional analysis of early B-lineage cells by in vitro S17 stromal cell assay, showing predominant B-lineage colony formation from Fr. A, but some myeloid generation from CLP stage cells. D: Functional analysis of early B-lineage cells by in vitro DL1-OP9 stromal cell culture, revealing significant T-lineage potential in Fr. A stage cells. |
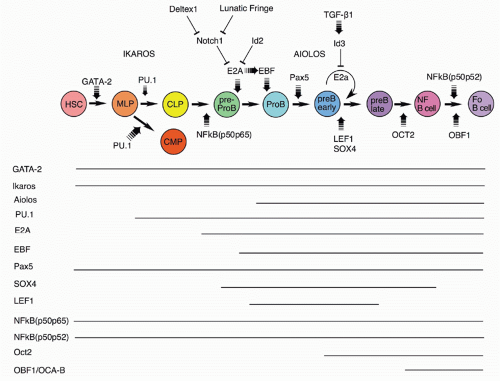 FIG. 8.4. Transcription Factors Important at Different Stages in B-Cell Development in Mouse Bone Marrow. Some regulatory networks are also shown. Positive/activating activity is indicated by arrows, whereas negative or blocking activity is indicated by bars. The rapidly cycling stage, early pre-B cell, is also indicated. Predominant stages of expression are indicated below the diagram. |
along the B lineage, as, while low-level expression induced in PU.1 null mice allowed B-lineage development, high-level expression blocked this and fostered myeloid lineage development,67 likely due to differential induction of the IL-7Rα and macrophage colony-stimulating factor receptor chains.68 In fact, retroviral mediated expression of the IL-7Rα chain complements defective B lymphopoiesis in PU.1 null bone marrow HPCs.69 Surprisingly, recent work from several groups indicates that some B-cell development can occur in the absence of PU.1 expression.70 Furthermore, analysis of conditional PU.1 knockout mice showed that expression of this transcription factor was not required after the pre-B-cell stage.71
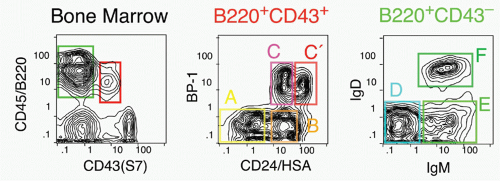 FIG. 8.5. Flow Cytometry Approach for Analyzing Different Stages of Cluster of Differentiation (CD)19+ B-Cell Development in Mouse Bone Marrow. Note that the antibody used for CD24/heat stable antigen (HSA) staining, 30F1, is important, as other monoclonal antibodies that recognize HSA do not resolve high from low-level expression as well. The cells expressing low levels of CD24/HSA, labeled “A”, are CD19- and is enriched for very early B-lineage precursors but also is contaminated by other cell types that can be detected by staining for AA4, NK1.1, DX5, and Ly6c; early B-lineage precursors are AA4+NK1.1-DX5-Ly6c-. |
to CD44 and VLA-4 on B-cell precursors, resulting in a disruption of normal pre-B proliferation in vitro.146 Such adhesion interactions may serve to transmit signals directly to the stromal cells or B precursors, or both. There is some evidence that stromal cells are induced to elaborate specific growth mediators after interaction with B-cell precursors or soluble regulators.147
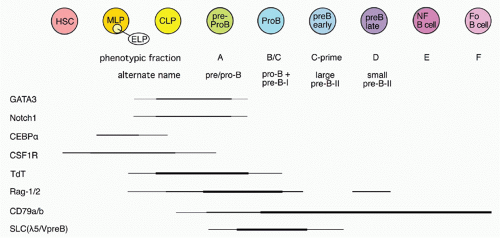 FIG. 8.6. Diagram of Distinct Phenotypic Stages and Characterization of Terminal Deoxynucleotidyl Transferase, Biphasic Rag Expression, Immunoglobulin-α/β, and Surrogate Light Chain Expression. Genes characteristic of myeloid and T-cell lineage are also shown. The cell type descriptions are cross-referenced to the alphabetic phenotypic fraction nomenclature and also to the Basel nomenclature. The early lymphoid progenitor population is identified by activation of the Rag-1 locus in a green fluorescent protein reporter mouse. |
IL-2, IL-4, IL-9, IL-15, and IL-21.154 IL-7Rα null mice have a severe deficit of both B and T cells in the periphery and lack most B-lineage cells in bone marrow.155 Mice with targeted inactivation of the γc or IL-7 do have some B-lineage development, suggesting an alternate cytokine; this appears to be thymic stromal lymphopoietin (TSLP). This protein was first identified as a pre-B-cell growth factor produced by a thymic stromal line156 and shows some of the same effects in culture as IL-7, although possibly inducing less proliferation and more differentiation.157 Its receptor has been cloned; it consists of two chains, the TSLP receptor and IL-7Rα.158 The TSLP receptor shares both sequence homology and genomic exon organization with the common gamma chain.159 Signaling through the IL-7 receptor requires JAK3 and activates the transcription factor STAT5, whereas signaling through TSLP is JAK3 independent but also activates STAT5.157,160 Unexpectedly, the growth response to TSLP requires synergy with the pre-BCR in bone marrow but not in fetal liver,161 leading some to propose that this might be a marker for distinctive B1 B-cell development in bone marrow.162 When TSLP is overexpressed from an inducible transgene, B1 B cells expand, apparently at the expense of marginal zone (MZ) B cells.163
TABLE 8.1 Regulators of Growth of Early B-Lineage Cells | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||
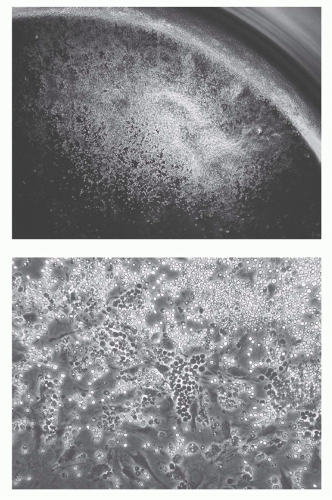 FIG. 8.7. Photomicrographs of B-Lineage Colony Proliferating on S17 Stromal Layer (in the Well of a 96-Well Microplate) in the Presence of Interleukin-7. Day 10 colony derived from a single Fr. A phenotype (see Fig. 8.5) cell. Low power and high power views. All of these cells now express CD19 and many have progressed to BP-1+. |
lineage.76 That is, expression of Notch1 by retroviral transduction has been shown to redirect B-lineage differentiation in bone marrow along the T lineage. Furthermore, a reciprocal result was found in conditional Notch1 null mice, blocking T-cell development in the thymus to be replaced by B-cell development.97 Finally, altering the Notch1 modifier lunatic fringe by overexpressing this molecule under regulation of an lck promoter resulted in B-cell development in the thymus.80 Differentiation of lymphoid precursors to NK or dendritic cell lineages was unaffected in Notch1 null CLP cells, so Notch apparently affects only the B/T-lineage decision.
through the BCR is mediated by accessory peptides, similar to the CD3 components of the T-cell receptor, known as Ig-α and Ig-β.224,225,226,227 Inactivation of Ig-β228 results in a block at the B220+CD43+ stage in mouse bone marrow, similar to that seen in µ-mt and λ5 null mice. Finally, the Syc tyrosine kinase plays a critical role in transducing BCR cross-linking signals in mature B cells and inactivation of this gene results in a “leaky” block at this same stage.229,230 Thus, any mutation that affects this pre-BCR complex (see following section; see Fig. 8.10A) precludes efficient progression past the earliest stages of B-cell development.
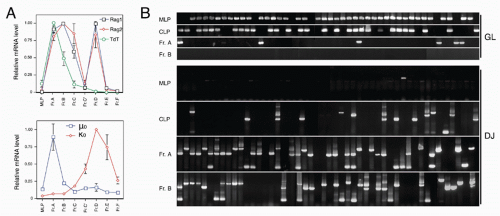 FIG. 8.8. A: Profiles of immunoglobulin (Ig) rearrangement-related gene expression. Cells isolated using fractionation scheme in Figure 8.3 for multilineage progenitors and Fr. A; in Figure 8.5 for Fr. B through Fr. F. Relative messenger ribonucleic acid levels assessed by performing semiquantitative reverse transcription-polymerase chain reaction using limited numbers of cycles, blotting, then probing, and quantitating the probe signal. Note the biphasic expression of Rag genes and the early expression of sterile µ (labeled µ0) during the first wave, where heavy chain rearranges, and the upregulation of sterile kappa transcripts (κ0) during the second Rag wave, when most light chain rearrangement takes place. B: Single-cell polymerase chain reaction analysis of Ig heavy chain rearrangement in four early stages of developing B-lineage cells. Deoxyribonucleic acid prepared from individual cells isolated following the scheme shown in Figure 8.3 was divided into two aliquots and analyzed for retention of a deoxyribonucleic acid segment lost upon any D-J rearrangement (labeled GL, germline) and also for D-J rearrangement. Note that D-J rearrangement initiates at the common lymphoid progenitor stage, where 30% to 50% of cells show a rearrangement. |
that pre-BCR signaling is more akin to “tonic” signaling in mature B cells.231,232,233 That is, simple assembly of the complex (or possibly some degree of multimerization fostered by the self-aggregating nature of SLC234) probably is sufficient for the cell to pass this developmental checkpoint (Fig. 8.10B). One clear-cut finding is that pre-BCR signaling in a transformed pro-B-cell model system can occur in the absence of any additional cell type, suggesting that if a ligand exists, it must be expressed on B-lineage cells rather than stromal cells.235 Possibly, pre-BCR homodimerization is mediated through glycosylation at a conserved asparagine residue in the first µ constant region domain.236 Studies of the T-cell analog of the pre-BCR, pre-Tα, provide strong evidence that it signals through spontaneous dimerization, without requirement for an external ligand.237,238,239
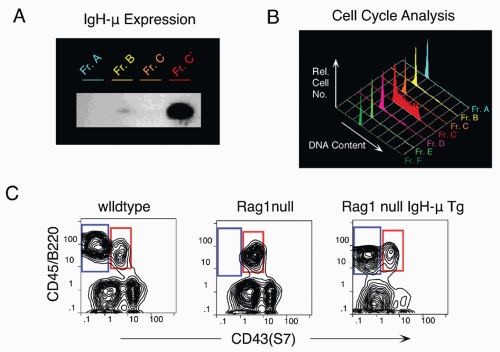 FIG 8.9. A: Western blot of immunoglobulin µ heavy chain expression showing high-level expression in Fr. C-prime. B: Cell cycle analysis of individual fractions shows most cells in Fr. C-prime are cycling. Propidium iodide staining of permeabilized sorted cells allows determination of deoxyribonucleic acid content per cell using flow cytometry. C: Block in B-cell development in Rag-1-deficient mice can be overcome by introduction of an immunoglobulin µ heavy chain transgene. |
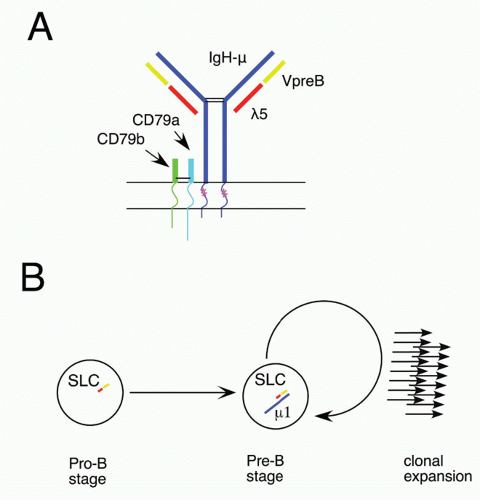 FIG 8.10. A: Diagram of the pre-B-cell receptor (BCR) µ heavy chain with surrogate light chain (λ5 and VpreB) in place of conventional light chain. As in the BCR, immunoglobulin (Ig)-α and Ig-β serve to couple signals between the receptor and cytoplasmic components, such as BLNK and Syk. Starred m transmembrane residues are important in mediating interaction with Ig-α/Ig-β, as mutation of these diminishes BCR function. B: Clonal expansion mediated by pre-BCR assembly. Association of newly generated µ heavy chain with pre-existing surrogate light chain leads to a burst of proliferation at the pre-B stage. |
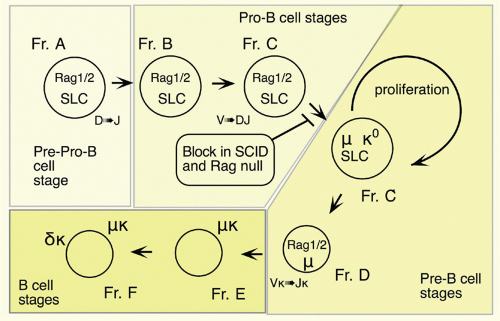 FIG. 8.11. Model of Mouse Bone Marrow B-Cell Development Showing Relationship of Immunoglobulin Rearrangement with Progression and Proliferation. |
IL-7 signaling, exits the cell cycle, and the Rag genes are reexpressed at high levels. Sterile kappa transcripts become detectable during the cycling stage, likely reflecting chromatin remodeling to make the kappa light chain locus accessible,252 so induction of the recombinase machinery can initiate kappa light chain V to J rearrangement.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


