idea that tissue destruction may lead to expanded immunopathology (nowadays referred to as “determinant spreading”) was proposed by Weil and Braun in 1907: In the course of an infectious disease, “tissue destruction results in the generation of antibodies that […] will be directed not only against the degraded products of the destroyed cells but also against human cell products as such. […] These autoantibodies could attack cells, [and] liberate antigen which in turn could induce the generation of autoantibodies.”13 Even the concepts of organ specificity,14 immunoprivilege, and a breakdown of regulatory mechanisms as a cause for autoimmunity7 were developed in the early days of autoimmunity research.
an infectious origin, diseases are called immunopathologically mediated, whereas lack of such knowledge results in reference to such diseases as autoimmune.41 While this argument is akin to the medical taxonomy where diseases of unknown origin are assigned to the domain of the “endogenous,” “idiotypic,” “essential,” or “primary,” a “positive” definition for autoimmune diseases is needed to provide a specific diagnostic framework that allows for unequivocal identification of distinct autoimmune disorders, yet remains flexible enough to accommodate new insights in etiologic and symptomatologic processes. A first attempt to provide such a basis for the establishment of the autoimmune origin of human diseases was formulated by Witebsky et al.42 who modeled their postulates on those of Koch: recognition of an autoimmune response (autoantibody or cell mediated), identification of a corresponding autoantigen, as well as induction of an analogous autoimmune response and disease in experimental animals. A timely update for these criteria has been proposed by Rose and Bona,43 who suggested a combination of direct evidence (transfer of pathogenic antibodies or T cells), indirect evidence (reproduction of disease in experimental animals), and circumstantial evidence (clinical clues) to determine an underlying autoimmune etiology for human diseases. However, it is important to note that any specific guidelines have to be tailored to individual autoimmune disorders. An example for a catalog of diagnostic criteria to be evaluated in a scoring system for identification of patients with a specific autoimmune disease is the report of the International Autoimmune Hepatitis Group.44 This report also illustrates the importance of distinguishing between an autoimmune and infectious origin for hepatitis44: Immunosuppressive therapy has a beneficial effect on the course of autoimmune hepatitis (AIH); responsiveness to such therapy is in fact one of the diagnostic criteria for AIH but may be detrimental when employed for treatment of virus-induced hepatitis.
determining the relative ranking of autoimmune diseases in terms of mortality risk among women under the ages of 65. Remarkably, the collection of 24 autoimmune diseases specified by Jacobson et al. ranked within the top 10 causes of death.48 Thus, autoimmune conditions not only decrease the quality of life among afflicted individuals but also due to their high prevalence constitute a major public health burden. In addition, a trend toward rising incidence rates among most autoimmune disease has been noticed over the past few decades. For example, the incidence of multiple sclerosis (MS) in Italy has doubled between 1981 and 2002,49 and in the United States, the incidence of celiac disease increased fivefold in 15 years.50 These trends are not likely to abate, and predictions for the number of new type 1 diabetes (T1D) cases in Europe are as high as 24,400 by 2020, whereas while the prevalence under the age of 15 years is estimated to rise from 94,000 in 2005 to 160,000 in 202051; in Finland, the country with the highest incidence of T1D, the number of new cases diagnosed at or before the age of 14 years will likely double in the next 15 years.52 The economic challenges associated with these developments are indeed staggering as shown by the health care-related expenses in the United States and provided by the American Autoimmune Related Diseases Association for Crohn disease ($10.9 to $15.5 billion in 2008), rheumatoid arthritis (RA; $19.3 billion in 2005), or psoriasis ($11.2 in 2010). Treatment costs are expected to increase even further due to the nature of some of the more succesful therapeutic modalities developed for chronic autoimmune conditions, for example, the introduction of antitumor necrosis factor (TNF) biologics. While clearly an important advance and of great benefit to patients, these drugs do not promote a cure for the underlying disease, and the need for continuous treatment will exacerbate associated health care costs.
TABLE 44.1 Prevalence of Autoimmune Diseases | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
the insulin-producing β cells in the pancreas.55,56 However, NOD mice also exhibit aspects of type 2 diabetes57 and are prone to autoimmune sialitis, thyroiditis, peripheral neuropathy, prostatitis, a lupus erythematosus-like syndrome that develops after exposure to killed mycobacteria, as well as, under certain circumstances, exocrine pancreatitis.56 Similar to the etiology of T1D, specific T cells are involved in the pathogenesis of all these disorders, although antigenic targets and requirements for costimulatory interactions are distinct. Thus, as in human T1D, the NOD mouse combines a generalized genetic susceptibility to multiorgan autoimmunity that is focused on pancreatic β cells but not limited to endocrine organs.
B cells can play important roles as APCs, and antibodies can possibly enhance disease pathogenesis. In both diabetes and MS, autoantibodies correlate with disease progression. In thyroiditis, autoantibodies are instrumental for causing disease and in AIH, their role is thought to be crucial.
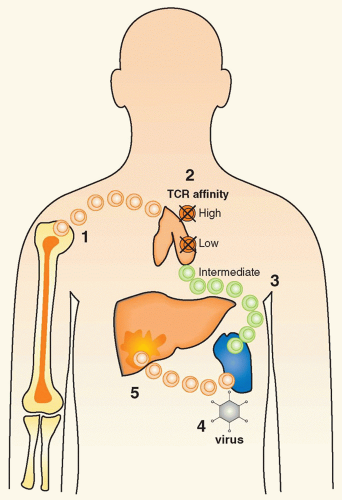 FIG. 44.1 Events Important for the Pathogenesis of Autoimmunity. 1: Lymphocyte precursors migrate from the bone marrow to the thymus to undergo maturation. 2: In the thymus of susceptible individuals, autoreactive lymphocytes can escape thymic selection. Whereas cells with high or low T-cell receptor affinity are generally deleted; “intermediate” lymphocytes fail to undergo negative selection. 3: Such initially naive cells migrate to peripheral organs such as the spleen and may remain unresponsive for many years. 4: Certain environmental triggers have the ability to activate these naive cells by bystander activation, molecular mimicry, or antigenpresenting cell cross-presentation. 5: As a consequence, activated autoreactive lymphocytes migrate to target organs such as the liver and mediate inflammation and tissue damage, leading to clinical autoimmune disease symptoms. |
attack, and lymphocytes detrimental in one organ/disease will not necessarily be detrimental to other organs, and ubiquitous autoimmunity will therefore occur very rarely.
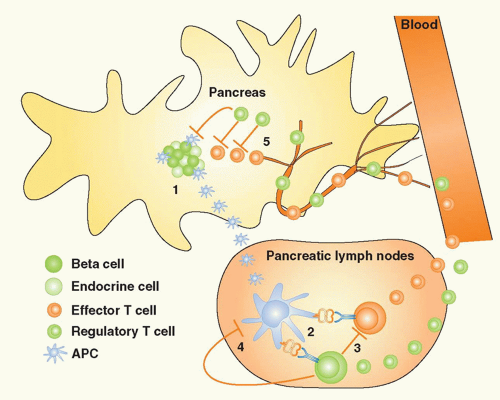 FIG. 44.2. The Concepts of Bystander Suppression and Infectious Tolerance. 1: Autoantigens are taken up by professional antigen-presenting cells (APCs) in the target organ, here depicted as the pancreas in type 1 diabetes. Upon maturation, APCs migrate to the draining lymph nodes. 2: In the lymph nodes, lymphocytes specific for distinct β cell antigens are activated by the APCs. 3: As a consequence, aggressive T cells directed toward one antigen could be regulated by regulatory T (Treg) cells specific for another autoantigen, a process termed “bystander suppression” by Howard Weiner.561 4: In theory, Tregs could also modulate APC function and foster induction of more Tregs to the same or other antigens, a process termed “infectious tolerance” by Hermann Waldman.562 5: Identical mechanisms may then regulate autoimmune responses in the target organ, after both effector cells and Tregs gain access via the circulation. |
that are not expressed during development, or were cryptic for an extended period of time, as well as antigens secreted and cross-presented in lymphoid organs by APCs, appear to be better suited for assuming the role as primary culprits.
infect target tissues and induce strong inflammatory responses and immune activation. While the association between viral infections and organ-specific autoimmune disorders is an intriguing possibility, it has been exceedingly difficult to demonstrate a causative role for specific viruses in human autoimmune diseases. Among the many obstacles are the fact that 1) all individuals undergo a multitude of viral infections during their lifetime; 2) one has to assume that viruses are frequently cleared at the time of diagnosis and viral footprints can be difficult to find in individuals affected by an autoimmune disease (“hit and run” event); 3) the precise viral strain, infection kinetics, and number of T cells and type of effector functions induced may play an instrumental role in determining its effect in an individual genetically at risk, necessitating a very detailed immunologic profiling139; 4) due to MHC polymorphism, there is a significant variation in specificity of the antiviral response; 5) viral infections might not, per se, trigger autoimmunity but affect an ongoing autoimmune process in a detrimental way; and 6) there now is increasing evidence that viral and other140 infections may rather prevent than enhance ongoing autoimmune responses, either by apoptosing aggressive T cells or by augmenting the Treg pool.141,142 Thus, a successful approach should be to first explore the underlying mechanisms in virally induced autoimmunity and then apply the precise insight and paradigms developed specifically to the human situation.
Molecular mimicry, which implies the cross-reactivity between viral and self-determinants as a principle cause or mechanism to enhance autoimmunity143,144
Bystander activation, which postulates that APCs and autoreactive lymphocytes will become activated indirectly as a consequence of the cytokine/chemokine by virus infection of a particular organ145
to develop less arthritis,162 and treatment with neutralizing antibodies to IL-17 or soluble IL-17 receptor alleviated joint inflammation.163 In humans, it was found that IL-17 is increased in RA sera and synovial fluid and is present in the T-cell rich areas of the synovium.164 Furthermore, IL-17 has profound inflammatory effects on a range of cell types within the joint. Several clinical trials are underway to either block IL-17 or the cytokines that govern the generation of Th17 cells such as IL-6.
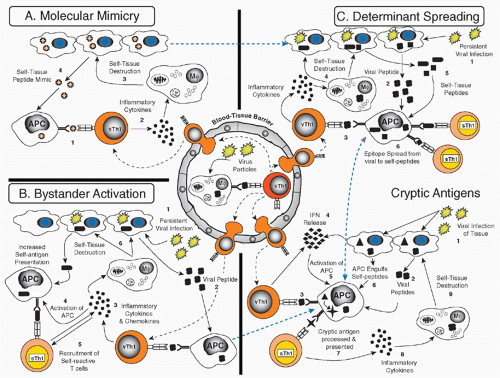 FIG. 44.3. Potential Mechanisms of Autoimmune Disease Induction. After a viral infection, activated virus specific T-helper cells (vTH1) and cluster of differentiation 8 cytotoxic T lymphocytes will migrate more readily through blood-tissue barriers to infected and noninfected organs. A: Molecular mimicry describes the activation of cross-reactive lymphocytes that recognize the original viral epitope and a self-epitope (1), which leads to the release of cytokines and chemokines (2), which enhance local inflammation, activate antigen-presenting cells (APCs), and indirectly or directly cause tissue destruction (3) and spreading of the autoimmune process (4). B: In the epitope spread model, persistent viral infections (1) could result in the activation of virus-specific T cells (2,3), which cause tissue damage by killing virally infected cells (4), leading to the release (5) and (cross-)presentation of more autoantigens (6). C: The bystander activation model describes the nonantigenspecific activation of autoreactive T cells. Infiltration by virally specific T cells (1,2) leads to inflammation and upregulation of immunity throughout the tissue (3,4), involving the activation of APCs, which now differentially process autoantigens (also de novo or previously cryptoic antigens). This can lead to activation of lymphocytes by T-cell receptor (TCR)-specific or TCR-independent mechanisms (5), which can then cause tissue damage (6). The cryptic antigen model describes the initiation of autoimmunity by differential processing of self-antigen/peptides, which can occur under inflammatory conditions. After viral infections, interferons (1) are secreted by antiviral T cells and infected cells (2-4). APCs are activated in this way (5), which enables them to engulf self-peptides (6, triangle) or to differentially process endogenous autoantigens. Cytokines can activate proteases more strongly, which might result in the presentation and processing of previously cryptic autoantigens (7-9). These displayed pathways are not mutually exclusive and are probably operational at different levels in many autoimmune responses. Currently of high interest is the presentation of neoantigens and strategies to define them. Courtesy of Steve Miller and Ludovich Croxford, Northwestern University, Chicago, IL. |
homeostatic baseline levels of immunity.88,90 In the absence of such regulatory mechanisms, immune responses will overshoot their goals and excessive immunopathology will occur. Thus, AICD is believed to play an important role in regulating autoimmunity. Apoptotic lymphocytes, for example, are easily detected in islet infiltrates in T1D,165 and targeted induction of limited apoptosis may even prevent onset of autoimmune disease.166 While increased apoptosis of aggressive lymphocytes that exceeds the “supply” of newly activated cells may directly limit an ongoing autoimmune process, limited apoptosis of target tissues may indirectly facilitate induction of protective regulatory responses. On the other hand, while apoptosis of target cells should at best be limited, decreased apoptosis of autoreactive aggressive lymphocytes will propagate autoimmunity. In RA, for instance, insufficient apoptosis of synovial macrophages, fibroblasts, and lymphocytes is one mechanism that might contribute to persistence of the disease and lead to synovial lining hyperplasia.167 It is therefore important to consider precisely which cells undergo apoptosis in order to predict the possible outcome.
If too many target cells die by apoptosis, organ destruction occurs more rapidly; however, at the same time, antigens releases from apoptotic cells appear to propagate tolerance rather than immunity.166,168
If Treg cells die by apoptosis, autoimmunity will be enhanced.169
If aggressive lymphocytes die by apoptosis, disease should be ameliorated. However, because they have to first be activated, they might induce organ damage during their activation phase.
of immunization in order to skew the resulting immune response to exert regulatory effector functions. Deletion of autoaggressive lymphocytes or anergy induction is even more risky, as only suboptimal immunization (ie, in the absence of costimulators) will result in this outcome. To control this in vivo is rather difficult. Ultimately, antigen-specific therapy will likely have to be individualized due to MHC polymorphism and distinct T-cell repertoires between individual patients and should be combined with other systemically acting agents, for example, antibodies against CD3 or CD4 or costimulation blockade. Indeed, there is intriguing evidence along these lines in animal models: Combination of a non-Fc-binding anti-CD3 antibody with mucosal immunization of insulin or insulin-derived peptides exhibited clear synergy and enhanced efficacy in reversing recent-onset diabetes in the NOD.177 In this case, induction of insulin specific adaptive Tregs and their effector function were enhanced and protection by these Tregs was transferable and highly effective in that it reversed recent-onset T1D.
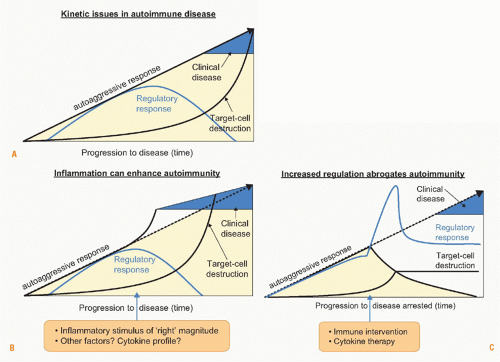 FIG. 44.4. Kinetic Issues in Autoimmune Diseases. A: Clinical manifestation of autoimmunity is the consequence of a dysequilibrium between protective (regulatory) and aggressive (effector) responses. An important consideration is that the destruction of the target cell or organ will usually lag somewhat behind the peak of the aggressive responses, because organ regeneration is common. B: Inflammatory stimuli will augment the effector arm of the autoreactoive response resulting in more rapid disease development. C: In contrast, induction of regulatory T cells can delay or dampen organ destruction. |
likely offer unique target sites for interventions with lower systemic side effects. However, one concern is that reestablishing proper immune homeostasis and regulation in one organ may still affect homeostasis systemically or at another site. Therefore, thorough preclinical evaluation and careful monitoring of undesired effects is urgently needed. Ideally, treatments have to be administered before complete organ destruction has occurred in patients identified by genetic or other screening to be at risk to develop full-blown clinical disease. During the preclinical state, frequently regulatory autoreactive responses are still strong and their augmentation can result in protection (Fig. 44.5). During advanced stages of autoimmunity, Treg function or susceptibility of effector T cells to regulation might decrease.182 The goal to reestablish homeostasis and proper regulation after an initial insult that caused organ-specific inflammation appears a natural countermeasure to which the specific immune system may be successfully harnessed. An intriguing example is the emerging concept of organ repair that can potentially be enhanced by drug therapy. In T1D, regeneration and replication of β cells can possibly occur after giving exenatide, a GLP-1 agonist, which is an established intervention in type 2 diabetes and under investigation in combination with other drugs such as anti-CD3 in T1D.
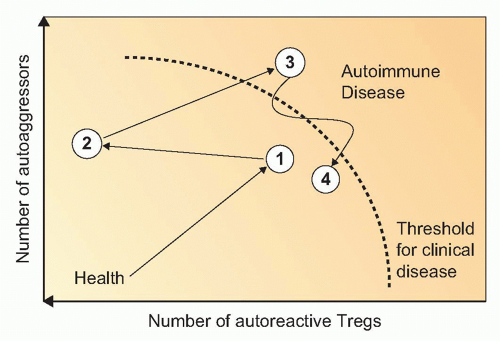 FIG. 44.5. Development of Autoimmune Disease as a Function of Aggression versus Regulation. Following induction of disease (1), distinct numbers of autoaggressive and autoreactive regulatory T cells will be generated and activated. Undulations between protective states and diseases states follow (2, 3) (ie, the so-called honeymoon phase in T1D), which can eventually lead to an irreversible state of clinical manifestation of disease (4). An additional important component to factor in is the ability of the target organ to regenerate. |
In other instances, direct virally induced thyroiditis has been documented epidemiologically with thyroid or parathyroid disease.
primary targets. The earliest islet cell-specifc antibodies in human individuals at risk are directed to insulin,227,228 and evidence from relevant animal models and human studies points toward insulin as a primary antigenic target,229 islet-specific glucose-6-phosphatase catalytic subunit related protein,230,231 and possibly glutamic acid decarboxylase (GAD),232 but definitive proof for a pathogenic role has only been ascertained in the NOD mouse model and not yet in humans.233 The cellular infiltrates found in the islets contain both CD4+ and CD8+ T lymphocytes and their irreducible role in β-cell destruction has been documented in several animal models.234,235,236 CD8+ T cells can exert direct cytotoxic effects toward MHC class I-expressing β cells, while CD4+ T cells secreting inflammatory cytokines and can provide help to CD8+ T cells as well as B cells. Ultimately, it is important to consider that β cells constitute about 60% of the islets, which in turn contribute only -2% to the pancreas mass and demonstrate, unlike many other tissues targeted in autoimmune disorders, an exquisite sensitivity to cytokines such as IL-1, TNF, and IFNs that will result in their apoptotic cell death after prolonged exposure.94
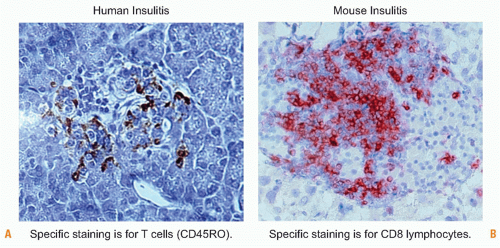 FIG. 44.6. Human and Mouse Insulitic Lesions. Comparison of human insulitis (Left), courtesy of Francesco Dotta (University La Sapienzia, Rome, Italy), and mouse insulitis (Right), from a rat insulin promoter-lymphocytic choriomeningitis virus mouse 14 days postinfection with lymphocytic choriomeningitis virus (von Herrath laboratory, La Jolla Institute for Allergy and Immunology, La Jolla, CA). |
processed peptide epitopes was similar, individuals with T1D produced more IFNγ relative to IL-10, whereas the ratio was the opposite in healthy individuals.
extended observation time involving infant as well as childhood consumption of cow’s milk. Therefore, a dietary link between milk feeding and T1D can be considered unlikely but not excluded after long-term exposure to cow albumin or other milk proteins. Similarly, wheat-derived gluten and milk-derived insulin have been implicated as a cause for childhood diabetes. The evidence, however, is not convincing at this point, and no firm links have been established.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


