FIGURE 7-1. Genes and polymorphic marker density of chromosome 8. The relative numbers of genes and SNPs are shown above chromosome 8. The microsatellite markers (short tandem repeats, STRs) and genes located within band q13 are shown below the chromosome. SNPs and microsatellites are essential for linkage and association studies. The gene structure of one of the genes in band 13, the paired box transcription factor PAX8, indicates that it consists of 10 exons. Alternative splicing of these exons generate multiple variants of PAX8.
From its very beginning, the development of ethical, legal, and social issues (ELSI) were important components of the HGP.17,18 The remarkably rapid discovery of new disease-causing genes and the advances in genetic testing continue to raise many ethical, social, and financial questions concerning the use of genetic techniques in medicine. For example, the discovery of the BRCA1 and BRCA2 genes, which predispose to breast and ovarian cancer, have enhanced our understanding of familial forms of these cancers, but how to best use genetic testing in potentially affected individuals and family members remains a challenging question. Breast cancer is a common disease, but mutations in the BRCA genes account for relatively few cases. Thus, the absence of mutation does not eliminate the risk of breast cancer; at present, it remains unclear how the presence of a mutation should be used in patient management. Should prophylactic mastectomy be performed, or should genetically susceptible individuals only undergo more intensive screening? What are the implications for insurability, employment, childbearing, and interpersonal relationships? The answers to these questions require additional clinical investigation and greater availability of genetic counseling.18 Protection against discrimination based on genetic information for health insurance and employment—for example, the recently introduced Genetic Information Nondiscrimination Act (GINA) in the United States—are important first steps to avoid misuse of genetic information.19 On the other hand, the discovery of the MEN2 gene has provided a useful strategy for identifying affected individuals with this highly penetrant autosomal-dominant disorder.3 In this case, unaffected individuals can be spared screening for pheochromocytoma, medullary thyroid cancer, and hyperparathyroidism. In carriers of a RET gene mutation, prophylactic thyroidectomy in early childhood permits these individuals to avoid the development of medullary cancer (see Chapter 90).
Categories of Genetic Disorders
Although many disorders are transmitted according to traditional Mendelian rules, it is now clear that a variety of different mechanisms can lead to genetic diseases (Table 7-2). Fundamental principles of genetic transmission are summarized briefly here, and additional information is available in other sources.20–22 Disorders of chromosome number or structure were among the first to be recognized because they can be detected using cytogenetic techniques. In endocrinology, disorders of the sex chromosomes, including Klinefelter syndrome (XXY) and Turner syndrome (XO), are particularly relevant. Molecular cytogenetics, in particular the advent of fluorescent in situ hybridization (FISH), has led to the identification of more subtle chromosome abnormalities such as microdeletions. The inheritance of either two maternal or paternal chromosomes, so called uniparental disomy, can be associated with endocrine disorders if it involves an autosome that is imprinted (see later).
Table 7-2. Mechanisms of Transmission of Genetic Endocrine Diseases
| Transmission | Example of Endocrine Disorder | |
|---|---|---|
| Gene | Disorder | |
| Chromosomal | XXY Multiple genes | Klinefelter syndrome |
| Autosomal-recessive | CYP21 (21-hydroxylase) | Congenital adrenal hyperplasia |
| Autosomal-dominant | CASR (calcium-sensing receptor) | Familial benign hypocalciuric hypercalcemia |
| X-linked | KAL1 (Kallmann) | Kallmann’s syndrome |
| Y-linked | SRY (testis-determining factor) | XY sex-reversal |
| MEN1 (menin) | Multiple endocrine neoplasia type 1 | |
| Mitochondrial | tRNA (Leu-UUR) | Diabetes-deafness syndrome |
| Mosaic | GNAS1 (Gsα) | McCune-Albright syndrome |
| Somatic | TSHR (TSH receptor) | Autonomous thyroid nodules |
| Imprinting | GNAS1 (Gsα) | Albright hereditary osteodystrophy |
| Multigenic | Multiple genes | Type 2 diabetes mellitus |
| Contiguous gene syndrome | Deletion of several genes | DiGeorge syndrome |
Mendelian disorders are caused by mutations in single genes. Information about these genetic disorders is available in the OMIM (Online Mendelian Inheritance in Man) database (see Table 7-1). The patterns of Mendelian transmission are now part of classical genetic teaching and include autosomal-recessive, autosomal-dominant, and X-linked disorders (Fig. 7-2). The transmission of genes or traits is typically depicted in family trees or pedigrees. Analysis of the pattern of transmission, particularly in large families with multiple generations, can be very valuable for predicting the mode of inheritance. This information is useful for genetic counseling, and it often narrows the differential diagnosis, particularly when mutations in several different genes can give rise to similar phenotypes (nonallelic or locus heterogeneity). For example, neurohypophyseal diabetes insipidus caused by mutations in the AVP-NPII gene is typically transmitted as an autosomal disorder.23 In rare cases, it can, however, be recessive.24 The nephrogenic form of diabetes insipidus can be X-linked due to mutations in the AVPR2 receptor, whereas mutations in aquaporin 2 are associated with a recessive or a dominant inheritance.25 For this reason, when dealing with a genetic disorder, it is important to obtain a detailed family history, often from several different family members. This information can then be combined with laboratory and genetic testing to arrive at an accurate diagnosis.
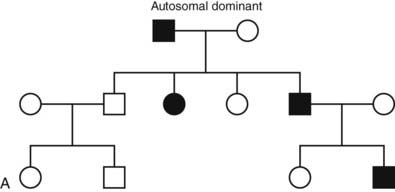
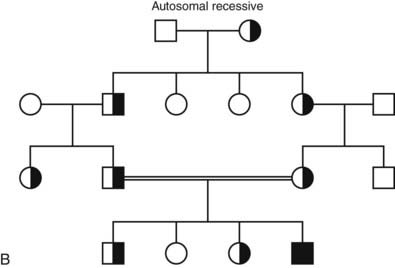
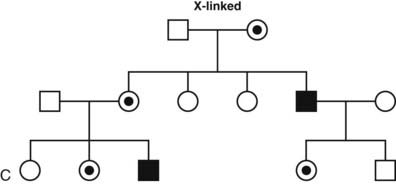
FIGURE 7-2. Classic patterns of Mendelian genetic transmission. A, Autosomal-dominant transmission. B, Autosomal-recessive transmission. C, X-linked transmission. Males are depicted by squares, and females are depicted by circles. Double lines linking parents indicate consanguinity. Affected individuals are shown by filled symbols. Half-filled symbols indicate heterozygous individuals. Dot-filled symbols indicate female carriers of X-linked traits.
AUTOSOMAL-DOMINANT DISORDERS
Diseases inherited in an autosomal-dominant manner are typically characterized by the presence of one mutant allele and a normal allele on the other chromosome. A single mutant allele is sufficient to cause the disorder. In some instances such as nonautoimmune familial hyperthyroidism, the gene is dominant because the mutations in the thyroid-stimulating hormone receptor (TSHR) are constitutively active26 (see Chapter 91). In other cases, such as thyroid hormone resistance, the mutant gene acts in a dominant negative manner to antagonize the function of the normal, wild-type gene27,28 (see Chapter 94). Mutations in one allele may be associated with haploinsufficiency, a situation in which a single normal copy provides insufficient protein to assure normal function (i.e., steroidogenic factor-1, SF1) 2008.28a Haploinsufficiency is a frequent mechanism of disease associated with mutations in transcription factors29 or rate-limiting enzymes.
In MEN1, a germline mutation in the tumor suppressor gene menin is transmitted in a dominant manner30 (see Chapter 150). If the second allele is inactivated by a somatic mutation, this will lead to neoplastic growth (Knudson two-hit mechanism). Whereas the defective allele in the germline is transmitted in a dominant way, the tumorigenic mechanism results from a recessive loss of the tumor-suppressor gene in affected tissues. Thus, the mechanisms by which genes act in a dominant manner are highly variable, even though they share similar features of transmission. In dominant disorders, the probability that an offspring will inherit the mutant gene is 50%, and individuals can be affected in each generation (see Fig. 7-2A). The disease does not occur in the offspring of unaffected individuals. Males and females are affected with equal frequency.
AUTOSOMAL-RECESSIVE DISORDERS
In an autosomal-recessive disease, both parents of an affected individual are obligate heterozygotes (see Fig. 7-2B). The affected individual, who can be of either sex, can be homozygous (inherit two copies of the same mutation) or inherit distinct mutations in each copy of the gene (compound heterozygote). Heterozygous carriers of a defective gene do not usually display phenotypic features of the disease. When both parents are heterozygous for a mutation, their offspring have a 25% chance of inheriting a normal genotype, a 50% probability of a heterozygous state, and a 25% risk of disease. If one parent is heterozygous and one is homozygous, the probability of disease increases to 50% for each child, and the pedigree analysis may mimic that of autosomal-dominant inheritance (pseudodominance). Most cases of homozygous mutations occur in situations of parental consanguinity or in isolated populations in which the gene pool is small. The likelihood of compound heterozygous mutations depends on the gene frequency in the population for each of the mutations, which is usually very low. Congenital adrenal hyperplasia caused by mutations in 21-hydroxylase is representative of autosomal-recessive disorders (see Chapter 103). There are many distinct mutations in the 21-hydroxylase gene (CYP21), and the prevalence of these mutations is high enough (~1/100 in most populations) that it is not unlikely for unrelated parents to be heterozygous. As a result, a child that inherits two distinct mutations in 21-hydroxylase will be affected with the disorder. Depending on the degree to which the mutation affects enzyme function, a range of phenotypic severity can be seen in different individuals.
X-LINKED DISORDERS
A daughter always inherits her father’s X chromosome, together with one of the two maternal X chromosomes. A son inherits one of the maternal X chromosomes and the Y chromosome from his father. Thus, there is no father-to-son transmission in X-linked inheritance, and all daughters of an affected male are obligate carriers of the mutant allele (see Fig. 7-2C). Because males have only one X chromosome, they are hemizygous for a mutant allele and are therefore more likely to develop the mutant phenotype. In females, the expression of X-chromosomal genes is influenced by X chromosome inactivation, which leads to random inactivation of most genes on one of the two copies. Occasionally, predominant X-inactivation of the normal allele can result in a partial phenotype in females carrying an X-linked trait (e.g., nephrogenic diabetes insipidus caused by AVPR2 mutations).25
Several endocrine disorders, including Kallmann’s syndrome, adrenal hypoplasia congenita, adrenal leukodystrophy, nephrogenic diabetes insipidus, androgen insensitivity, and hypophosphatemic vitamin D-resistant rickets, are transmitted in an X-linked manner. As expected from the aforementioned mechanism of X-linked transmission, these disorders are much more common in males than females.
Y-LINKED DISORDERS
The Y chromosome carries a small number of genes, and there are few Y-linked disorders. One of the Y-chromosomal genes, the sex region–determining Y gene (SRY), which encodes the testis-determining factor (TDF), can cause XY sex reversal when mutated.31 Alternatively, translocation of the SRY gene to the X chromosome can cause an XX male phenotype. Another group of genes on the Y chromosome includes the highly repetitive DAZ genes that are important for spermatogenesis. (Micro)deletions of these genes, often transmitted as a new germline mutation, are an important cause of azoospermia and male infertility.32,33
Relationship Between Genotype and Phenotype in Genetic Disorders
Variations in the clinical phenotype in inherited disorders are common and can be explained by various mechanisms.
Allelic heterogeneity indicates that multiple different mutations can occur in the same gene. In some instances, there is a clear genotype-phenotype correlation between a specific allele and the phenotype. Certain mutations can completely inactivate a protein, whereas others retain partial function. For example, the phenotype of androgen insensitivity includes a wide spectrum of disorders that ranges from severe resistance and testicular feminization to partial resistance in Reifenstein’s syndrome.34 Allelic heterogeneity is often a problem for genetic testing, because one must often examine the entire gene to definitively exclude a mutation.
Nonallelic or locus heterogeneity designates a situation in which a similar disease phenotype results from mutations in different genes. For example, the nephrogenic forms of diabetes insipidus can be caused by mutations in the X-chromosomal arginine vasopressin receptor 2 gene (AVPR2), whereas mutations in the aquaporin 2 gene (AQP2) cause either autosomal-recessive or autosomal-dominant nephrogenic diabetes insipidus.25 Nonallelic heterogeneity can pose problems for genetic testing since different genes may have to be considered along with the possibility of allelic heterogeneity in each of the candidate genes.
A phenotype not caused by inheritance of a mutated gene that is identical or similar to a genetic trait is called a phenocopy. For example, goiter may be the consequence of defects in thyroid hormone synthesis, or it can be the consequence of nutritional iodide deficiency.35
Sometimes there are marked phenotypic differences in individuals carrying the same mutation. If some individuals harboring the mutation fail to express the phenotype, the trait is said to display incomplete penetrance. Expressivity is used to describe the phenotypic spectrum in the individuals with the disorder. Expressivity is thus dependent on penetrance. The phenotypic variation leading to incomplete penetrance and variable expressivity can be explained by environmental factors, modifier genes, or sex. The modification of the effect of genes through gene-gene interaction is commonly referred to as epistasis. Incomplete penetrance in some individuals leads to skipping of generations and can confound pedigree analysis. Variable expressivity and penetrance illustrate that genetic and/or environmental factors may influence “simple” Mendelian traits. This has practical relevance for genetic counseling because one cannot always predict the course of disease even when the mutation is known.
Aside from mutations in the sex chromosomes, some diseases are expressed in a sex-limited manner because of the differential function of the gene product in males and females. For example, gain-of-function mutations in the LH receptor cause dominant male-limited precocious puberty in boys because activation of the receptor induces testosterone production in the testis, whereas it is functionally silent in the immature ovary.36
Variations to Simple Mendelian Inheritance Patterns
Many diseases display a familial clustering without having a clear pattern of classic Mendelian inheritance (see Table 7-2). This applies to the complex disorders, which are underlying the pathogenesis of many congenital defects and major health care problems such as type 2 diabetes mellitus, hypertension, obesity, osteoporosis, heart disease, and psychiatric disorders. These disorders are thought to involve multiple different genes, each of which contributes partially to the disease phenotype, either directly or through gene-gene interaction(s). Since the contribution of any one these genes is usually relatively weak, they are difficult to localize using classical genetic linkage approaches in large pedigrees. Therefore, the genetic analysis of complex disorders relies heavily on large population-based genomewide association studies (GWAS).8,37,38 In this approach, one can search for genetic variants that occur with increased frequency in affected individuals. Alternatively, one can search for genetic variants that occur more often in affected sib-pairs versus the population at large. These analyses are challenging because of the large number of individuals who need to be studied (several hundreds to thousands). In addition, the entire genome must be searched for candidate genetic markers associated with the disease, which is now easily performed with comprehensive bead- or microarray-based genotyping of single nucleotide polymorphisms (SNPs) (see Fig. 7-1, Fig. 7-3). Because many different genes (and environmental events) contribute to the pathogenesis of these disorders, this type of gene search is most successful when one can identify specific subphenotypes that are likely to be caused by a relatively small number of genes, or by studying relatively homogenous populations.39 For example, the sib-pair approach has been used successfully for the identification of genetic loci associated with type 1 diabetes mellitus,38 and association studies have identified loci that are involved in insulin secretion and action in the Amish population.39 A relatively large number of GWAS have now validated known genes and identified novel genes and loci associated with diabetes mellitus type 2.40
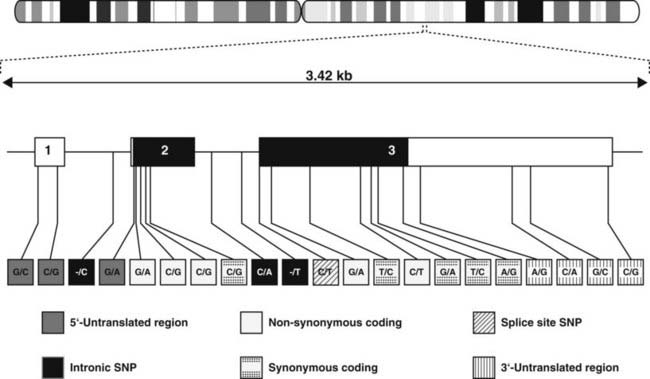
FIGURE 7-3. Single-nucleotide polymorphisms. Single-nucleotide polymorphisms in the TRH gene, which encodes the thyrotropin-releasing hormone. The gene is located on chromosome 3q13.3-q21. The transcription of this gene, which contains three exons, occurs from left to right. SNPs are found in all regions of the gene, the 5′-untranslated region, the exons, splice sites, introns, and the 3′-untranslated region. In the coding region, the SNPs may be synonymous (i.e., the encoded amino acid remains unchanged) or nonsynonymous (i.e., the alteration results in an amino acid substitution). SNPs are found about every 300 bp throughout the genome.
Each mitochondrion contains several copies of a circular chromosome which encodes proteins that are part of the respiratory chain, and transfer and ribosomal RNAs. The mitochondrial genome does not recombine and is transmitted exclusively through the maternal line, since all mitochondria reside within the oocyte cytoplasm. Thus, mitochondrial disorders are only transmitted from mother to offspring, and males and females are affected equally. Mitochondrial disorders typically have complex clinical features that often involve muscle and brain because of the high dependence of these tissues on oxidative phosphorylation. Moreover, these disorders often have endocrine manifestations. For example, the maternally transmitted diabetes-deafness syndrome is due to mutations in the mitochondrial gene encoding the tRNA for leucine.41
In addition to the inactivation of one of the two X chromosomes in females (X-inactivation), gene inactivation also occurs on selected chromosomal regions of autosomes.42 This phenomenon, referred to as genomic imprinting, leads to preferential expression of an allele depending on its parental origin and can influence the expression of certain genetic diseases. The classical examples involve the Prader-Willi syndrome and the Angelman syndrome, which are caused by genes located on the short arm of chromosome 15. In the Prader-Willi syndrome, deletions are found exclusively on the paternally-derived chromosome. Alternatively, Prader-Willi syndrome may be caused by the inheritance of two maternal copies of chromosome 15, that is, maternal uniparental disomy 15. In contrast, patients with Angelman syndrome have deletions in the same region of chromosome 15, but they either occur only on the maternally-derived chromosome or they have paternal uniparental disomy 15.43 Genomic imprinting is also involved in the various clinical presentations associated with mutations in the GNAS1 gene encoding the stimulatory Gsα subunit (see Chapter 65).44 Heterozygous loss-of-function mutations in the GNAS1 gene lead to Albright hereditary osteodystrophy (AHO). Paternal transmission of GNAS1 mutations leads to the AHO phenotype alone (pseudopseudohypoparathyroidism) (Fig. 7-4), whereas maternal transmission leads to AHO in combination with resistance to hormones such as parathyroid hormone (PTH), thyroid-stimulating hormone (TSH), and gonadotropins, all of which stimulate G protein–coupled seven-transmembrane domain receptors (pseudohypoparathyroidism type IA). These phenotypic differences are due to a tissue-specific imprinting of the GNAS1 gene, which is expressed primarily from the maternal allele in tissues such as the thyroid, gonadotropes, and the proximal renal tubule. In most other tissues, the GNAS1 gene is expressed biallelically. In patients with isolated renal resistance to PTH (pseudohypoparathyroidism type IB), an imprinting defect of the GNAS1 gene results in decreased Gsα expression in the proximal renal tubules.44
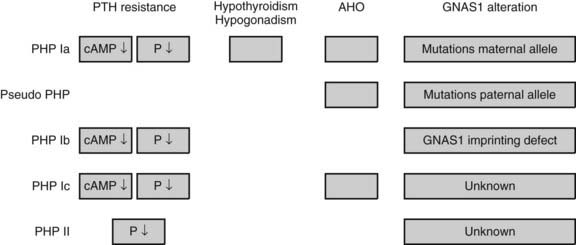
FIGURE 7-4. Effects of imprinting of the GNAS1 gene. The GNAS1 gene encodes the stimulatory Gsα subunit. Inactivating mutations cause Albright hereditary osteodystrophy (AHO). Maternal transmission leads to AHO in combination with resistance to hormones such as PTH, TSH, and gonadotropins, which all stimulate G-protein seven transmembrane receptors (pseudohypoparathyroidism type IA, PHP IA). Paternal transmission of GNAS1 mutations leads to the AHO phenotype alone (pseudopseudohypoparathyroidism). These phenotypic differences are due to a tissue-specific imprinting of the GNAS1 gene, which is expressed primarily from the maternal allele in tissues such as the thyroid, gonadotropes, and the proximal renal tubule. In most other tissues, the GNAS1 gene is expressed biallelically. In patients with isolated renal resistance to PTH (PHP type IB), an imprinting defect of the GNAS1 gene results in decreased Gsα expression in the proximal renal tubules.44 The molecular basis of PHP Ic and PHP II remain to be elucidated.
Mosaicism refers to the presence of two or more cell lines in an individual that differ in their genotype. Mosaicism can result from a mutation that occurs during embryogenesis or later in development. The developmental stage at which the defect arises will determine whether germ cells or only somatic cells are involved. Somatic mosaicism is characterized by a patchy distribution of somatic cells containing a mutation. For example, activating mutations that occur in the Gsα subunit early in development cause the McCune-Albright syndrome.45 The clinical phenotype, which can include ovarian cysts that secrete sex steroids and cause precocious puberty, polyostotic fibrous dysplasia, café-au-lait skin pigmentation, pituitary adenomas, and hypersecreting autonomous thyroid nodules, varies depending on the tissue distribution of the mutation.
Somatic mutations also play an important role in various forms of neoplasia.46,47 Comprehensive genomewide analysis of cancers with high-throughput sequencing technologies has led to the detection of point mutations in novel cancer genes and to the recognition that some cancer genomes carry numerous somatic rearrangements.47 When somatic mutations enhance cell proliferation or prolong cell survival, they can be associated with the clonal expansion of the cell population and the development of tumors. Activating mutations in the TSHR or Gsα can cause autonomously functioning thyroid nodules (see later). In inherited cancer syndromes in which the “first hit” has already been transmitted in the germline, somatic mutations in the second allele of the involved tumor suppressor genes play an important role (Knudson two-hit model). MEN1 provides an example of such a disorder.
Trinucleotide repeats are found in several genes, and their number varies among healthy individuals (polymorphic variants). For example, the number of CAG repeats found in the first exon of the androgen receptor (AR) gene is lowest in African Americans, intermediate in Caucasians, and highest in Asians.48 An increase in the number of repeats above a certain critical threshold, however, is associated with the X-linked form of spinal and bulbar muscular atrophy and partial androgen resistance (SBMA, Kennedy syndrome).49 Several other trinucleotide disorders are frequently associated with endocrine features. For example, male patients with dystrophia myotonica frequently present with hypogonadism,50 and the risk for developing diabetes mellitus correlates with the length of the repeat expansion in patients with Friedreich’s ataxia.51
Principles of Genetic Linkage and Association
Genetic linkage refers to the fact that genes are physically attached to one another along the length of the chromosome. Consequently, two genes that are close together on a chromosome are usually transmitted together, unless a recombination event separates them. Recombination, which occurs during meiosis, is useful for purposes of mapping genes, because it provides a landmark that delineates borders for the location of a gene. The odds of a cross-over, or recombination event, are proportionate to the distance that separates them. Thus, genes that are far apart are more likely to be separated by a recombination event than genes that are close together. These features make it possible, given large pedigrees or populations, to calculate the genetic distance between two genes. A centimorgan (cM) is defined as a recombination frequency between two loci of 1% and corresponds to about 1 Mb of DNA (see Fig. 7-1).
Linkage is usually expressed as a logarithm of the odds (lod) score, which is a ratio that reflects the probability that the disease and marker loci are linked rather than unlinked.52 Positive numbers favor linkage and negative scores support nonlinkage. Lod scores of +3 are generally accepted as supporting linkage, whereas a score of −2 is consistent with the absence of linkage. When candidate genetic regions have been identified by linkage, more detailed analyses can be performed using additional markers, or if the region is small enough, one can attempt to identify the disease gene among the many genes present within a particular locus.
The presence of polymorphic variation in DNA is essential for linkage studies. Genetic variation provides (1) a means to distinguish the maternal and paternal chromosomes in an individual and (2) markers of different regions along the chromosomes. Historically, these polymorphisms consisted of restriction fragment length polymorphisms (RFLPs), in which nucleotide sequence variation altered the presence of specific restriction sites in DNA. Thus, when combined with Southern blot analysis, RFLPs allow one to track the transmission of genes within a pedigree. Although principles of RFLP analysis are useful for understanding disease transmission and gene mapping, the technique has been supplanted by other means of polymorphic analysis. Initially, short tandem repeats (STRs) or microsatellites consisting of highly repetitive 2-, 3-, or 4-bp sequences have been used for linkage studies. The human genome project has generated high-density maps of STRs throughout the entire genome (see Fig 7-1.). Now, genotyping of SNPs has replaced the characterization of STRs. A SNP is a single base-pair variation at a specific locus, and it forms the most common form of genetic variation. SNPs occur roughly every 300 bp and are found in noncoding and coding regions (see Fig. 7-3). SNPs within a coding sequence can be synonymous (i.e., not altering the amino acid code) or nonsynonymous. There are roughly 3 million differences between the DNA sequences of any two copies of the human genome. Comprehensive genotyping of up to a million SNPs in the genome of an individual can now be performed easily with microarray and bead array technologies, an approach that is essential for GWAS.
After the identification of the approximately 10 million SNPs that are commonly found in the human genome, the International HapMap Project has generated a catalog of common genetic variants that occur in individuals from distinct ethnic backgrounds (see Table 7-1).53 SNPs that are in close vicinity are inherited together as blocks referred to as haplotypes, hence the name HapMap. These haplotype blocks can be identified by genotyping selected SNPs, so-called Tag SNPs, an approach that reduces cost and workload (Fig. 7-5).
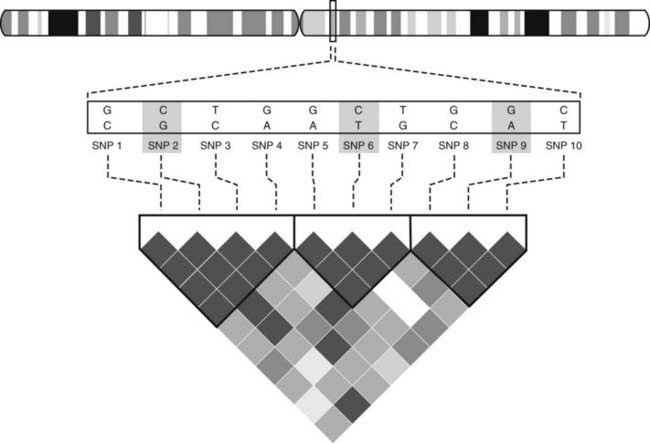
FIGURE 7-5. Haplotype blocks. The figure shows 10 SNPs in the selected genomic region. The associations between these SNPs has been determined and is shown graphically. Dark grey indicates the strongest association, white the lowest association. This analysis reveals that three blocks of haplotype blocks occur in this region. In order to characterize the region through genotyping, one can rely on so-called Tag SNPs (SNP 2, 6, 9) as representatives of the three blocks, rather than genotyping all SNPs. Such information is available for the whole genome and for different ethnic groups, and it permits performing genomewide association studies (GWAS) more efficiently and at lower cost.
Allelic association designates a significantly increased or decreased frequency of an allele with a disease. This can be due to a true biological association or linkage dysequilibrium, that is, association due to close linkage. Association studies compare a population of affected individuals with a control population—for example, affected individuals and matched controls or affected and unaffected siblings.37,40 Allelic association studies are useful for identifying susceptibility genes in complex disorders. Association studies have revealed a role for the HLA region, the insulin VNTR (variable number of tandem repeats), and the CTLA4 (cytotoxic T-lymphocyte-associated 4) gene in diabetes mellitus type 1 (see Chapter 40). In diabetes mellitus type 2, associations have been established with a variety of genes: TCF7L2 (a transcription factor regulating the insulin and glucagon genes), PPARG (a nuclear transcription factor that is a target of glitazones), KCNJ11 (a potassium channel expressed in β cells), and SLC30A8 (a zinc transporter) (see Chapter 41).38,40
Methods Used to Detect Gene Deletions and Point Mutations
Mutations, any change in the primary nucleotide sequence, are structurally diverse. They can affect one (point mutations) or a few nucleotides, consist of gross numerical or structural alterations in individual genes or chromosomes, or involve the entire genome. Mutations may be located in regulatory regions, introns, or exons. Point mutations occurring in the coding region may lead to amino acid substitutions (missense mutations), premature stop codons (nonsense mutation), frameshifts, or they can be silent. Mutations in introns can introduce or destroy sequences that are important for proper splicing of the precursor RNA to the mature mRNA.
Recombinant DNA approaches that are used to investigate a particular disorder are usually based on clues derived from its clinical and pathophysiologic characteristics, which in some cases allow one to predict the gene that harbors a defect. For example, it is reasonable to postulate that selective growth hormone (GH) deficiency in several members of a family might be due to a defect in the gene encoding GH, GH-releasing hormone (GHRH), or the GHRH receptor. In other cases, however, there are no obvious candidate genes. For example, in the MEN syndromes, it was difficult to predict a gene that causes proliferation of selected lineages of endocrine cells. In this case, the most expeditious approach was to localize chromosomal regions carrying markers associated with the disease phenotype through linkage studies (see earlier). After candidate genes were identified, they could be analyzed for mutations in affected patients to verify that they cause the disorder.
DETECTION OF MUTATIONS
Previous editions of this chapter have discussed several techniques used for the detection of mutations in more detail.5,10 Analyses of large alterations in the genome are possible using cytogenetics, fluorescent in situ hybridization (FISH), Southern blotting, high-throughput genotyping, and sequencing. More discrete sequence alterations rely heavily on the use of the polymerase chain reaction (PCR).54 The PCR technique is a very powerful tool for molecular diagnostics for the following reasons. First, the dramatic amplification of DNA allows diagnostic analyses using very small amounts of initial starting tissue. Sufficient DNA for PCR is routinely extracted from lymphocytes or from cells present in saliva, hair, amniotic fluid, chorionic villi, or other accessible tissue sources. Second, because PCR uses short synthetic oligonucleotides to prime the reaction, it can be readily applied to any known sequence. PCR is also essential for linkage and association studies because highly polymorphic sequences (microsatellites and SNPs) can easily be amplified.55
In many cases, the PCR is the starting point for more detailed characterization of a mutation. In early studies to identify a specific mutation, the PCR product is often used for DNA sequencing to establish the location and base change that is present in the gene. Because the amount of DNA provided by PCR is relatively large, automated DNA sequencing methods allow direct sequencing of the DNA fragment without an intervening subcloning step.56 Alternatively, the amplified DNA fragment can be subcloned into a plasmid vector to allow subsequent DNA sequencing. Direct sequencing has the advantage that the sequence analysis is based on a large population of amplified DNA molecules rather than individual clones that may contain PCR-generated sequence errors (which occur in approximately 1/3000 bases). Direct DNA sequencing of the PCR product also has the advantage that a heterozygous mutation can be detected by the presence of two different nucleotides at the mutant position.
After characterization of a specific mutation or SNP in an index patient, other family members or individuals can be easily screened for the presence or the absence of the mutation with a variety of techniques such as direct sequencing, allele-specific PCR, TaqMan PCR, RFLP analysis, denaturing gradient gel electrophoresis (DGGE), single-stranded conformational polymorphism (SSCP), denaturing high-performance liquid chromatography (D-HPLC), oligonucleotide-specific hybridization (OSH), and mass spectrometry (see previous edition).5,10
HIGH-THROUGHPUT SEQUENCING TECHNOLOGIES
Several high-throughput sequencing platforms (also referred to as next-generation or massively parallel sequencers) have become commercially available. The advent of these technologies is rapidly changing the scope of genetic and genomic analyses. They provide the possibility to sequence very large regions and entire genomes at a relatively low cost. This allows identification of disease-causing mutations and characterization of SNPs at high resolution. Other applications include the characterization of RNA expression, noncoding and microRNAs, protein-DNA interactions, epigenomic alterations, and metagenomic analyses.57 High-throughput sequencing processes millions of sequence reads in parallel rather than 96 reactions at a time as on current capillary systems. The workflow to produce next-generation sequences consists of the following main steps. Following the creation of a library with fragments of the template DNA, which requires only a few micrograms of starting material, these fragments are ligated to specific adaptor oligos to both ends of each DNA fragment. These single-strand fragments are then amplified and sequenced using different technologies (pyrosequencing, polymerase-based sequencing by synthesis, ligation-based sequencing), depending on the platform. High-throughput sequencers produce much shorter read lengths (35 to 250 bp, depending on the platform) than capillary sequencers (650 to 800 bp). The enormous amount of data generated by a single run on a high-throughput sequencer requires intense computational capacity and sophisticated bioinformatics in order to assemble the sequence and to make comparisons to one or several reference sequences.58
FUNCTIONAL STUDIES OF MUTANT HORMONES AND RECEPTORS
Identification of a DNA sequence alteration is not sufficient to establish that it is responsible for the disease phenotype. First, nucleotide substitutions could represent polymorphisms or DNA sequence variations that occur in the population as a whole. If the base change occurs in the coding sequence but does not alter an amino acid, it is most likely a polymorphism. Occasionally, however, a seemingly silent mutation may affect mRNA stability or splicing mechanisms.59 If a mutation results in an alteration of the amino acid sequence, it is still possible that the amino acid substitution is physiologically silent and does not significantly alter protein function. One approach for addressing the issue of polymorphisms is to screen a large number of normal individuals (e.g., 100) for the putative mutation. If the amino acid substitution is found in normal individuals, it is by definition a polymorphism, although it may have subtle functional consequences. Substitution of codons that are highly conserved across species (presumably implying functional importance) have a higher chance of being mutations than amino acids that are more variable in different species. Finally, mutations tend to be linked to the disease phenotype when examined in several family members, whereas polymorphisms should sort randomly unless they are located close to the disease locus. Although these determinations of polymorphisms are not entirely reliable, they are of practical importance because the next steps of assessing the functional importance of a candidate mutation can require significant experimental effort.
Assuming there is evidence against a polymorphism, it is almost always possible, using recombinant techniques, to assess the effect of a mutation in a functional assay. As illustrated in Fig. 7-6, recombinant mutant and wild-type proteins can be expressed and subjected to a variety of functional assays. In the example shown, expression of mutant luteinizing hormone β (LHB) allows it to be assessed for its ability to form an α-β heterodimer, to undergo glycosylation, to bind to its receptor, and to activate receptor signaling pathways.60 Mutant enzymes such as 21-hydroxylase can be analyzed for their ability to bind substrate or to carry out catalysis.61 Mutant receptors like the insulin receptor have been subjected to an array of functional tests, including insulin binding, receptor autophosphorylation, receptor internalization, and receptor stability.62 Likewise, G protein–coupled receptors (GPCRs) containing activating or inactivating mutations can be tested for their abilities to bind hormones and to signal through second messenger pathways.26,36,63 Nuclear receptors such as the thyroid hormone receptor can be analyzed not only for their ability to bind thyroid hormone and DNA target sequences but also for their capacity to function as transcription factors in transient gene-expression assays.64 These types of studies establish whether a given mutation is of functional importance and provide insight into protein structure/function and hormone action. In many instances, these experiments of nature provide rapid identification of critical functional domains because identification of the mutations is biased by the presence of a recognizable clinical phenotype.
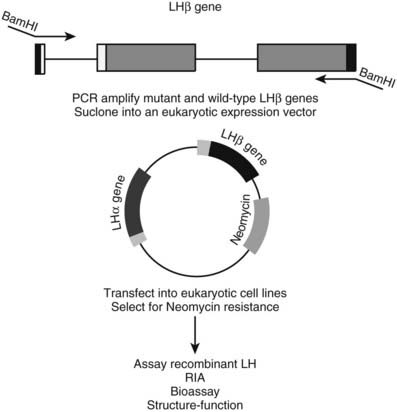
FIGURE 7-6. Expression of recombinant hormones to verify the biological effect of a mutation. The ability to express recombinant hormones or receptors is an important strategy for testing the effects of putative disease-causing mutations. A variety of expression systems are available, including E. coli, baculovirus, vaccinia virus, and mammalian cells. The choice of expression system is dictated by a variety of issues, including characteristics of the protein, desired production level, and the nature of the bioassay that will be used. A strategy for producing recombinant LH is illustrated for the purpose of analyzing the functional consequences of mutations in the LHβ gene.60 The LHβ gene is amplified by PCR from genomic DNA and inserted into a eukaryotic expression vector. The presence of a viral long terminal repeat (LTR) provides a strong promoter to drive expression in transfected mammalian cell lines. Because LH is a heterodimer containing an α and β subunit, the vector is designed to contain a copy of the α gene to allow the genes to be coexpressed from the same plasmid. A third gene encoding neomycin resistance allows selection and isolation of clonal cell lines that have been successfully transfected with the expression vector. Secreted LH can be analyzed by structural methods and for its functional properties. This approach allows delineation of hormone domains important for α-β subunit dimerization, receptor binding, and receptor activation.
Overview of Inherited Endocrine Disorders
Molecular biology has already transformed the practice of endocrinology and will continue to do so.65 In addition to improving our understanding of hormone and receptor structure/function, these techniques allow the production of large quantities of recombinant insulin with or without altered kinetics, GH, gonadotropins, thyrotropin, and erythropoietin, among others. For many monogenic disorders, molecular testing is now available and has a growing impact on clinical management (see Table 7-1, Table 7-3). Genetic testing has an established role in the evaluation of families with MEN23 and in hereditary and some forms of sporadic pheochromocytoma.66,67 Lastly, the current “postgenomic” disciplines will undoubtedly lead to the identification of unknown molecules involved in endocrine systems and provide novel insights into metabolic networks.68,69
Table 7-3. Approach to the Patient With a Suspected Genetic Disease
DGGE, Denaturing gradient gel electrophoresis; D-HPLC, denaturing high-performance liquid chromatography; SSCP, single-stranded conformational polymorphism.
APPROACH TO THE PATIENT
Careful clinical evaluation, in combination with selected ancillary biochemical and imaging studies, remains the first step in unraveling the underlying pathogenic mechanisms of a disease.7 The family history is of great importance to recognize the possibility of a hereditary component. A detailed pedigree is extremely useful for the recognition of hereditary traits, the assessment of genetic risk, and for purposes of genetic counseling.70 Because of the possibility of age-dependent expressivity and penetrance, the family history often needs continuous updating. In general, genetic testing should only be performed after obtaining informed consent. The patient and the family should be carefully informed about the potential consequences of positive results which may carry psychological distress and the possibility of discrimination. In the case of a positive genetic test result, the physician must discuss the potential implications for the affected individual and other family members. The patient must be informed that it is his or her responsibility to contact relatives who should undergo testing because the physician is prohibited from contacting family members without their formal permission. Equally important is discussing the meaning of a negative result, which should include an explanation of false-negative or inconclusive results and technical limitations of the tests.
Genetic testing in children poses distinct ethical issues.71 In most instances, it should be limited to situations in which it has an impact on the medical management. If there is no apparent benefit, testing should be deferred until the patient can consent independently.
Laboratories performing genetic tests can be easily identified through the GeneTests web site (http://www.genetests.org/servlet/access), an international laboratory directory (see Table 7-1). For certain rare disorders, the test may only be available through research laboratories. If a disease-causing mutation is expected in all cells as a result of germline transmission, DNA can be collected from any tissue, such as nucleated blood or buccal cells, for cytogenetic and mutational analyses. In the case of somatic mutations, which are limited to the neoplastic tissue, an adequate sample of this lesion is required for the extraction of DNA or RNA.
GENETIC ENDOCRINE DISORDERS
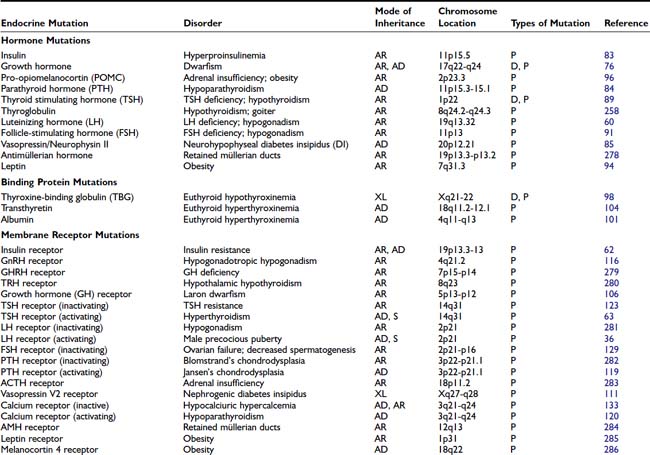
Several hundred endocrine disorders exhibit an inheritance pattern suggestive of a primary gene defect (see OMIM and Human Gene Mutation Database).72,73 Gene mutations have now been identified in many of these disorders (Table 7-4). Some common themes emerge even from the genetic defects that have been described to date. First, the phenotypic variability that characterizes many endocrine diseases is often reflected in genetic heterogeneity. Some clinical phenotypes that were thought previously to represent distinct diseases can now be interpreted as manifestations of different types of mutations within a single gene (e.g., the clinical variants of congenital adrenal hyperplasia can be attributed to distinct mutations in 21-hydroxylase or other enzymes involved in steroid biosynthesis).74 Second, the propensity of certain genes to be frequent targets for mutations may be explained in part by gene structure and organization. Genes such as GH that have been duplicated to form gene clusters are predisposed to undergo recombination and deletion.75,76 Third, although many of the mutations reported initially have been associated with severely affected patients, it is now obvious that mutations with less severe consequences can result in a more discrete phenotype and that there is often a phenotypic spectrum. For example, the phenotype of androgen insensitivity includes a spectrum of disorders that ranges from severe resistance in the case of testicular feminization to milder resistance in Reifenstein’s syndrome and other syndromes of mild androgen resistance associated with gynecomastia and infertility.34 These disorders are each caused by mutations in the androgen receptor, but the mutations result in different degrees of receptor dysfunction. In some cases, the receptor is deleted or mutated in a manner that it is completely inactive. In other examples, mutations perturb the amount or stability of the receptor, causing partial resistance. Phenotypic variation can also be ascribed to environmental influences and/or the action of other genes called modifier genes. A further extension of this concept is that genetic polymorphisms (DNA sequence variants) in the normal population cause subtle differences in hormone or receptor activity, thereby constituting part of the basis for the variability that is seen in the normal range of hormone levels and activity.

Because a staggering number of mutations in different endocrine genes has already been reached,5,6,65 it is not practical to describe each of these disorders in a comprehensive manner. The interested reader is referred to individual chapters and to the references in Table 7-4. In addition, these disorders are described in the OMIM, the Human Gene Mutation, and the GeneTests databases (see Table 7-1). It is nevertheless useful to provide an overview of mutations that occur at different steps in endocrine pathways to illustrate the breadth and heterogeneous nature of disorders caused by gene defects.
HORMONE MUTATIONS
One might have expected that mutations in hormones would represent a common molecular basis for endocrine disorders. This does not appear to be the case, however. For the most part, causes of hormone deficiency syndromes remain enigmatic. For example, GH deficiency rarely involves deletions or mutations in the GH gene.77 Rather, most cases can be attributed to an inherited or acquired hypothalamic defect that can involve GHRH, the GHRH-producing neuron, the GHRH receptor, or one of the regulatory pathways that control GHRH secretion.78 GH deficiency is reminiscent of idiopathic hypogonadotropic hypogonadism, which is not due to a GnRH gene defect but rather to a defect in several genes (KAL1, FGFR1, PROKR2, PROK2, CHD7, FGF8) that control migration of the GnRH-producing neurons.79
The GH gene is a member of a large gene cluster that also includes a GH variant gene as well as several structurally related chorionic somatomammotropin genes and pseudogenes (highly homologous but functionally inactive relatives of a normal gene). Because such gene clusters contain multiple homologous DNA sequences arranged in tandem, they are particularly prone to undergo recombination, leading to gene duplication or deletion. It has been proposed that mispairing of areas with sequence homology can lead to unequal crossover during meiosis, with resultant gene duplication on one chromosome and gene deletion on the other chromosome.76,80 In autosomal-recessive isolated GH deficiency IA (IGHD IA), deletions of the GH gene represent one of the best-studied hormone mutations77 (see Chapter 26). Southern blot analyses of DNA from affected children demonstrate homozygous deletions of the GH gene, consistent with the autosomal-recessive inheritance pattern for transmission. Recessive point mutations in the GH gene lead to IGHD IB. In IGHD II, autosomal-dominant mutations in the GH gene encode a misfolded protein that aggregates with the normal GH protein, thus exerting a dominant-negative effect.81,82 These complexes may also be toxic to somatotrope cells.
Other mutations that have been described in hormones are listed in Table 7-4. The mutations in preproinsulin prevent processing of the preproinsulin precursor molecule, causing secretion of biologically inactive insulin molecules.83 A mutation in the signal sequence of PTH causes hypoparathyroidism, even when only one of the two PTH alleles is affected.84 It has been shown that this mutation interferes with hormone transport and processing, leading to the hypothesis that the mutant molecule could interfere with the transport of other cellular proteins, including the normal PTH protein. Most mutations in the AVP-NPII gene, which encodes vasopressin and neurophysin, appear to be somewhat analogous to the PTH mutation. An autosomal-dominant form of diabetes insipidus is caused by heterozygous mutations in the signal peptide or the neurophysin moiety of the AVP-NPII precursor protein.85 These amino acid changes in the signal sequence and carboxy-terminal vasopressin carrier protein, neurophysin, suggest that abnormalities in protein processing may prevent vasopressin synthesis or result in cellular toxicity. In vitro studies demonstrate abnormal processing and cellular toxicity of the mutant forms of vasopressin,23,86 and mice carrying one mutated allele show progressive loss of arginine vasopressin (AVP)-producing neurons relative to oxytocin-producing neurons,87 explaining the delayed onset of the disease.88 Very rarely, mutations lead to amino acid substitutions in the AVP moiety. In this case, the disease is inherited in a recessive manner.24
Homozygous mutations in the TSHB gene cause hypothyroidism. One TSH β-subunit mutation defines a region of the molecule that is required for heterodimerization with the α subunit,89 whereas others either truncate the protein or interfere with its biological activity.90 An LH β-subunit mutation defines a region that is critical for binding to the LH receptor.60 Interestingly, follicle-stimulating hormone β (FSHB) mutations have different phenotypes in males and females. They cause primary ovarian failure in females because of a defect in follicular maturation and estrogen synthesis.91,92 In males, FSHB mutations do not impair virilization or testosterone production, but they cause variable impairment of spermatogenesis.93 Mutations in leptin, initially described in murine models of obesity, have now been identified in humans.94,95 Unexpectedly, mutations in POMC not only cause adrenal insufficiency but also lead to obesity, apparently because of a role for α-melanocyte-stimulating hormone (MSH) in appetite control96 (see Chapter 27).
In these and other autosomal-recessive disorders, there is an opportunity to examine the effect of a gene knockout in humans. Thus, elimination of functional hormones such as GH, LH, FSH, or TSH allows one to attribute specific physiologic roles to the hormone that may be difficult to discern in normal individuals. For example, complete elimination of LH with retention of normal FSH allows the functions of these hormones to be distinguished. Creation of gene knockouts and knockins by homologous recombination in mice, or transgenic overexpression, allow the creation of animal models for studying hormone and receptor function (see Chapter 2).
BINDING PROTEIN MUTATIONS
The binding protein mutations cause little in the way of clinical disease, but if not recognized, they often lead to unnecessary treatment. As shown in Table 7-4, these defects are predominantly confined to proteins that bind thyroid hormone (see Chapter 93). Thyroxine-binding globulin (TBG) is the major thyroid hormone transport protein in the serum. Abnormalities in its production are not rare, occurring in approximately 1 in 2500 male births.97 Complete TBG deficiency is X-linked and is clinically apparent in males who are euthyroid but have very low levels of thyroid hormone in the serum. They are often mistakenly treated for hypothyroidism. Although large deletions of the TBG gene have not been found,98 a number of different point mutations can cause functional loss of TBG, either by alterations in protein structure or glycosylation.97 TBG excess is most commonly due to effects of drugs or hormones, although familial cases with amplification of the TBG gene have been reported.99 In the latter situation, the total TBG and thyroxine levels are elevated three- to fivefold in hemizygote males and two- to threefold in heterozygote females.
In addition to TBG, thyroid hormone binds to albumin and transthyretin (TTR). Familial dysalbuminemic hyperthyroxinemia (FDH) is an autosomal-dominant condition in which albumin has an increased affinity for thyroid hormone.100 Most cases are caused by a mutation in codon 218, in which there is replacement of arginine by histidine101 or proline.102 It is characterized by elevated levels of total thyroxine (T4) and normal levels of free T4 and TSH. Another form of the disorder causes a selective increase in triiodothyronine (T3) binding.103 The presence of abnormal binding proteins can be detected using electrophoretic analyses of serum thyroid hormone binding proteins or, now, by mutational analyses. Occasionally, TTR can be overproduced by pancreatic endocrine tumors, or mutant forms may have increased affinity for T4.104 The impact of TTR mutations on T4 affinity is variable and can be normal, decreased, or increased. Many of them are associated with an autosomal-dominant form of amyloidosis, affecting predominantly the peripheral nervous system and the heart.105
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


