Fig. 2.1
Structures of major bisphosphonates used clinically
Bisphosphonates have been used since the 1970s across the whole spectrum of bone resorption disorders , including Paget’s disease and cancer-related bone destruction. Their exact molecular mechanisms of action are now better understood. Within bone, bisphosphonates are internalized selectively by osteoclasts and interfere with specific biochemical processes. The antiresorptive effects on osteoclasts of the nitrogen-containing BPs (including alendronate , risedronate , ibandronate , minodronate, and zoledronate ) appear to result principally from their strong and selective inhibition of farnesyl pyrophosphate synthase (FPPS), a key enzyme in the mevalonate pathway of cholesterol biosynthesis. This pathway generates isoprenoid lipids utilized for the posttranslational modification (prenylation) of small GTP-binding proteins that are essential for osteoclast function.
The inhibition of farnesyl pyrophosphate synthase (FPPS) in the mevalonate pathway results in the accumulation of the upstream metabolite, isopentenyl pyrophosphate (IPP) , that may be responsible for immunomodulatory effects on gamma delta (γδ) T cells. Accumulation of IPP can also lead to production of another ATP metabolite called ApppI, which has intracellular actions, including induction of apoptosis in osteoclasts. BPs may have other biologically important cellular effects on inhibiting osteoclast differentiation, on decreasing tumor cell viability, and on preventing osteocyte apoptosis, the latter possibly through other pathways, e.g., connexin channels [8].
The pharmacological effects of BPs as inhibitors of bone resorption appear to depend upon two key properties: their affinity for bone mineral and their inhibitory effects on osteoclasts [9]. There are differences in binding affinities for bone mineral among the clinically used BPs, which may influence their distribution within bone, their biological potency, and their duration of action. Although it is obvious that different BPs share many pharmacological properties, it is also obvious that every BP has a specific and often unique profile. Clinicians may understandably question whether these pharmacological differences are of practical importance. Based on the available data, we have proposed that these differences may be clinically relevant [10]. Thus, there is evidence to indicate that there may be differences among the BPs in terms of the speed of onset of fracture reduction, efficacy at different skeletal sites, and the degree and duration of reduction of bone turnover, which may influence how long to treat patients with individual drugs (Fig. 2.2).
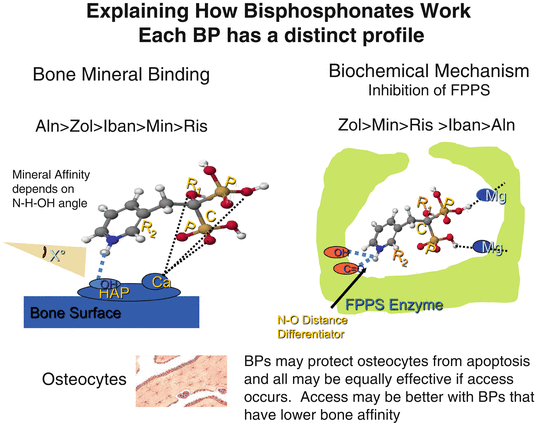
Fig. 2.2
Explaining how bisphosphonates work. They differ in their binding affinity for bone mineral and their inhibitory activity on farnesyl pyrophosphate synthase (FPPS). These properties contribute to the pharmacological potency and differences among the bisphosphonates
Bisphosphonates are currently the leading drugs used worldwide for the treatment of osteoporosis . In randomized controlled trials (RCTs) , alendronate, risedronate, and zoledronate have shown to reduce the risk of vertebral, non-vertebral, and hip fractures, whereas RCTs with ibandronate show anti-fracture efficacy at vertebral sites. The clinical effects of bisphosphonates on fracture reduction derived from RCTs and other studies are reviewed by Papapoulos in Chap. 15.
There are however difficulties with comparing the clinical effects of individual bisphosphonates with each other and with other antiresorptive drugs, because of the lack of direct comparisons in head-to-head clinical trials with appropriate endpoints (e.g., fracture ). Despite this, using the data generated from individual clinical studies, there is evidence for potential differences among BPs in terms of their clinical effects on speed of onset of fracture protection, sites of anti-fracture efficacy, and duration of effect. This may reflect their different profiles in terms of mineral binding properties and biochemical actions within cells.
Apart from the “big” four ( alendronate, risedronate, zoledronate, ibandronate), other bisphosphonates also have been used in osteoporosis but have not achieved broad indications and been licensed for this indication. These include clodronate, pamidronate, tiludronate, neridronate, minodronate, and olpadronate. Etidronate was approved in most countries in the early 1990s. Pamidronate has been extensively used off-label for osteoporosis as it was the only intravenous bisphosphonate available before zoledronate. It is still used in the management of osteogenesis imperfecta [11].
In the case of osteoporosis, some of the currently debated topical issues include deciding whom to treat and for how long, which BP to use, and how to manage poor compliance [12]. In general, BPs have proved to be not only highly effective but also very safe drugs. Nonetheless, issues of side effects and adverse events attract often disproportionate attention, as with osteonecrosis of the jaw (ONJ) and atypical femoral (subtrochanteric) fractures (AFFs), where the nature of any association with BPs remains unclear [13]. This is discussed in other chapters in this book.
Bisphosphonates have now become largely generic drugs, as key patents have expired, but are likely to remain major drugs for treating bone diseases for some time to come. Advances continue to be made, such as producing formulations to overcome interference of intestinal absorption by food [14]. Novel bisphosphonates have been made with even greater potency and selectivity, but they will have to have added benefits if they are to compete with cheaper generics. In looking ahead, there are obvious opportunities for extending the use of BPs to other areas of medicine. Several recent studies suggest that BPs may be associated with other clinical benefits outside the field of bone diseases, e.g., on mortality, cardiovascular disease, and reduction of colon cancer. The pharmacology underlying these potential effects needs to be understood. Further intriguing examples of non-skeletal effects include inhibition of several protozoan parasites, increasing longevity in animal progeroid models, and enhancing human stem cell life span, DNA repair, and tissue regeneration. Several of these effects may be explained by modulation of the isoprenylation of proteins that have regulatory functions in many cell types. We may be entering an era in which BPs are viewed as modulators of mevalonate metabolism (“MMMs”) , rather than as bone-active drugs. This research is opening up a wide range of new potential medical uses of bisphosphonates, including effects on T cells, tissue regeneration, radioprotection, and extension of life span.
Denosumab
Denosumab is a fully human monoclonal antibody against RANK ligand (RANKL) marketed under the name of Prolia® (Pralia® in Japan) for osteoporosis and Xgeva® for the prevention of skeletal complications in oncology. The discovery of the essential role of RANK ligand and of RANK signaling in osteoclast differentiation, activity, and survival led the way to developing denosumab. Denosumab acts by binding to and inhibiting RANKL from binding to RANK, leading to the loss of osteoclasts from bone surfaces.
RANK ligand is one of the two cytokines that are essential and sufficient to induce osteoclast differentiation. The other is the macrophage colony-stimulating factor (M-CSF). M-CSF binds to c-Fms, a single transmembrane domain receptor of the tyrosine kinase family, and RANKL binds to RANK, a single transmembrane receptor of the tumor necrosis factor (TNF) receptor family, which forms trimers upon ligand binding. The role of M-CSF is to first activate the proliferation and survival of cells of the monocyte–macrophage lineage and the expression of RANK, allowing the action of RANKL, which, together with M-CSF, constitutes an absolute requirement for commitment to and progression of early precursors along the osteoclast lineage. The differentiation step from osteoclast precursors to multinucleated osteoclasts is then induced by RANKL, again through the M-CSF-dependent expression of RANK at the cell surface of these early precursors. M-CSF and RANKL are both secreted by bone marrow stromal cells and osteoblasts, whereas RANKL is also secreted by T cells and to a lesser extent by B cells. The other component of the RANKL/RANK system is osteoprotegerin (OPG), which is a shed extracellular portion of the RANK receptor . Most of the cells producing RANKL also produce this decoy RANK receptor, OPG, which acts as an antagonist to RANK signaling and osteoclastogenesis by scavenging RANKL in the extracellular environment. OPG is also a regulated molecule, and it is the ratio between RANKL and OPG which determines the level of activation of RANK and therefore the extent to which osteoclast production and function is activated. All these molecules are thought to act in a paracrine manner and to regulate bone resorption locally. The overall endocrine regulation of skeletal homeostasis involves interactions between systemic hormones and the RANKL/RANK/OPG system. The expression and secretion of both RANKL and OPG are regulated by several calcium-regulating hormones, including estrogens, parathyroid hormone (PTH), and vitamin D3.
The first attempts to develop therapeutics based on these pathways utilized a chimeric OPG-Fc fusion protein to antagonize RANKL. However, the formation of neutralizing antibodies against OPG after administration of the fusion protein and its potential cross-reactivity with tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) led to the more attractive strategy of inhibiting RANKL directly using denosumab.
The key pharmacological differences between denosumab and the bisphosphonates reside in the distribution of the drugs within bone and their effects on precursors and mature osteoclasts [15]. This may explain differences in the degree and rapidity of reduction of bone resorption, their potential differential effects on trabecular and cortical bone, and the reversibility of their actions.
In the FREEDOM pivotal phase 3 clinical trial, denosumab was shown to significantly reduce vertebral, non-vertebral, and hip fractures by 68 %, 20 %, and 40 %, respectively, compared with placebo. In the FREEDOM study, 7808 women with postmenopausal osteoporosis were randomized to receive denosumab 60 mg subcutaneously once every 6 months or placebo [16]. FREEDOM is the acronym for the Fracture Reduction Evaluation of Denosumab in Osteoporosis every 6 Months study.
These and further studies involving over 12,000 patients have confirmed the efficacy and overall safety of denosumab [17–20], out to 8 years of treatment in the ongoing extension studies. Denosumab treatment increased BMD at the total hip, lumbar spine, and/or femoral neck and reduced markers of bone turnover to a significantly greater extent than oral bisphosphonates in women who had not received bisphosphonates at all or in the recent past or in those who had switched from alendronate to denosumab treatment.
There are several respects in which the differences between bisphosphonates and denosumab have practical consequences. It appears that the effects of denosumab to increase BMD continue on prolonged treatment, whereas they may plateau earlier on bisphosphonates.
Denosumab may also have a greater impact on protecting cortical bone than bisphosphonates, which is likely to contribute to its efficacy in reducing fractures.
However, the effects of denosumab rapidly reverse when treatment stops which means that ensuring compliance is even more important if potential benefits are to be sustained. Another difference is the potential use of denosumab in patients with impaired renal function, in whom use of bisphosphonates is contraindicated.
Estrogens : Background and Current Status
The recognition that the decline in estrogen production at the menopause was associated with bone loss dates back to the time of Albright more than half a century ago. The use of estrogens as HRT (ERT or hormone replacement therapy) in postmenopausal women was therefore a logical approach toward alleviating the symptoms and consequences of the menopause. After the link between menopause and osteoporosis was first identified, estrogen treatment became the standard means for preventing bone loss, even though there was no fracture data. This changed dramatically when results from the Women’s Health Initiative (WHI) study were published, which showed an increase in heart attacks and breast cancer [21]. Even though the risks were small, this prompted a large drop off in estrogen use. In later analyses, the WHI study showed that estrogen reduced fractures and actually prevented heart attacks in the 50–60-year age group. Estrogen alone appeared to be safer to use than estrogen plus the progestin medroxyprogesterone acetate and actually reduced breast cancer. The overall benefits and risks of estrogens are now better appreciated and are being continuously reappraised [22, 23]. In one analysis of hysterectomized women, it was estimated that by avoiding estrogen, there had been ∼50,000 extra deaths from 2002 to 2011 [24]. Currently, it is widely accepted that estrogens are effective in relieving menopausal symptoms and in preventing perimenopausal bone loss, as well as having other potential benefits, but their use as first-line therapy in osteoporosis is no longer appropriate.
The way in which estrogens act on bone and indeed on other tissues is complex. Estrogens may have direct effects on bone cells, but many of their effects may be indirect, mediated, for example, by reducing the actions of the RANK-ligand system on osteoclast differentiation. Estrogens exert their cellular effects mainly by binding to estrogen receptors (α & β), which are members of the large family of nuclear hormone receptors that regulate the transcription of specific genes. The different cofactors present in various tissues result in differential effects of individual ER ligands in key target tissues [25].
Estrogens can also exert rapid cellular responses by non-genomic mechanisms by binding to another type of receptor, called the G protein-coupled estrogen receptor 1 (GPER), formerly referred to as G protein-coupled receptor 30 (GPR30). GPER is an integral membrane protein with high affinity for estradiol and is a member of the rhodopsin-like family of G protein-coupled receptors.
Selective Estrogen Receptor Modulators
Selective estrogen receptor modulators (SERMs) are a diverse group of naturally occurring or synthetic nonsteroidal compounds that exhibit tissue-specific estrogen receptor (ER) agonist or antagonist activity, i.e., they can act as estrogens on some tissues but antiestrogens on others. The biochemical basis for differential actions on various target tissues is most likely to be due to the different conformation changes induced in the receptor after binding to individual ligands. These conformational changes in the ER in turn result in different patterns of binding to various coactivators and corepressor molecules , thereby leading to distinctive patterns of gene transcription and protein expression in the various target tissues.
The pharmacological profile of individual SERMs is therefore determined by the effects observed on the key target tissues that are important for human health. An ideal SERM would therefore have positive effects on the cardiovascular system and bone , without stimulating breast or endometrial tissue and raising the risk of cancer. Attempts to meet this ideal profile have been partially successful with currently approved SERMs such as raloxifene and bazedoxifene. However, it has so far proved impossible to wean out the adverse effects of provoking hot flushes or venous thrombosis from any of the individual SERMs in current clinical use. There are several comprehensive reviews of estrogens and SERMs available [26, 27].
Many different molecules with estrogen agonist or antagonist activity have been synthesized and evaluated experimentally. Several have been examined for their clinical potential. Some have failed to progress on account of unacceptable side effects or lack of desired efficacy. These include droloxifene, idoxifene, and arzoxifene . Overall, there have been more failures than successes in clinical development. There are currently only two SERMs approved for use of osteoporosis, namely, raloxifene and bazedoxifene (Fig. 2.3).
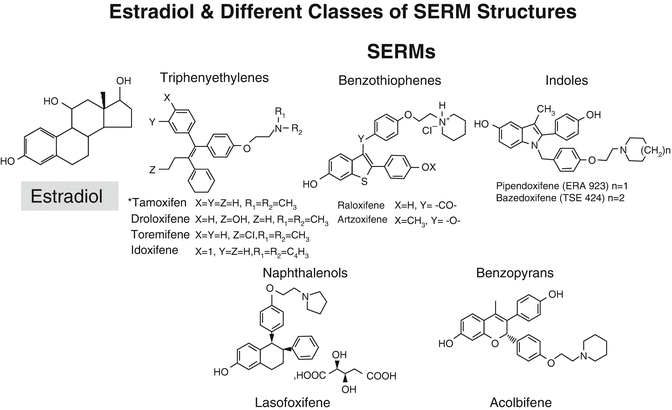
Fig. 2.3
Structures of estradiol and of various SERMs. It is interesting to note how many of the SERMs have two –OH groups separated by a similar distance to that found in estradiol, which contributes to their binding to the estrogen receptor (ER)
The earliest SERMs developed for clinical use were agents such as clomiphene , still used for induction of ovulation, and tamoxifen used as an antiestrogen in the management of breast cancer. Currently, other FDA-approved SERMs include toremifene, also used for prevention and treatment of breast cancer, and ospemifene approved for treatment of dyspareunia from menopausal vaginal atrophy. Raloxifene is approved for the prevention and treatment of osteoporosis and prevention of invasive breast cancer. Bazedoxifene is also approved in many countries either as a single agent but interestingly also as a combined preparation with estrogen. This tissue-selective estrogen complex (TSEC) involves a pairing of conjugated equine estrogens with bazedoxifene and is approved by the FDA. This pairing is designed to reduce the risk of endometrial hyperplasia that can occur with the estrogenic component of the TSEC without the need for a progestogen in women with a uterus. The combination also allows for the estrogen to control hot flushes and to prevent bone loss without stimulating the breast or the endometrium.
Although bisphosphonates remain first-line therapy for most patients, SERMs such as raloxifene and bazedoxifene provide a safe and effective alternative to bisphosphonates for women who are unable to tolerate bisphosphonates and for younger women with an increased risk of fracture who may remain on therapy for many years [28, 29].
Raloxifene
The first SERM developed specifically for use in osteoporosis was raloxifene , which interestingly had also being previously studied by Lilly but discontinued for its potential as an anti-breast cancer agent. Raloxifene is an estrogen agonist in bone and the liver. It increases BMD and reduces LDL cholesterol. It does not stimulate the endometrium and is a potent antiestrogen in the breast. There have been many clinical studies with raloxifene. In the large Multiple Outcomes of Raloxifene Evaluation (MORE) trial, a total of 7705 women who had a mean age of 65 years (low bone mass group) and 68 years (osteoporosis group) were randomly assigned to raloxifene (60 or 120 mg) or placebo daily and were followed up for a total of 8 years. There were significant increases in BMD at the spine and hip, and the risk of vertebral fracture was significantly reduced by 30 and 50 %, respectively, with raloxifene at 60 or 120 mg/day compared with placebo. There was no effect on non-vertebral fractures, but importantly treatment with raloxifene reduced the incidence of invasive and invasive estrogen receptor-positive breast cancers by more than 60 %
Bazedoxifene
Bazedoxifene is a novel synthetic “third-generation” estrogen agonist–antagonist, with an attractive experimental and clinical profile, with apparent added advantages when used in combination with conjugated estrogen (20 mg bazedoxifene plus 0.45 mg CE).
Several clinical studies have been reported, showing increases in BMD at the spine and also at the hip. In the pivotal phase III treatment study of bazedoxifene, 7492 healthy postmenopausal women (average age, 66 year) were randomly assigned to daily oral doses of bazedoxifene 20 or 40 mg, raloxifene 60 mg, or placebo. There was a reduction in spine fractures on all doses at 3 years of 42 % for bazedoxifene at 20 mg, 37 % for the 40 mg dose, and 42 % for raloxifene at 60 mg [30]. The efficacy of bazedoxifene was sustained through 5 and 7 years of treatment as observed in the extensions of the core trial [31].
Bazedoxifene reduced non-vertebral fractures when results from both doses were combined, and this became more evident when higher-risk groups were studied. A recent analysis of the data using FRAX® indicates that reductions in non-vertebral fractures appear to be greater than with raloxifene in the higher-risk groups [32].
Bazedoxifene treatment was not associated with detectable effects on risk of breast cancer, or cardiovascular outcomes, but had positive effects on lipid profiles. Bazedoxifene had neutral effects on the reproductive tract and was not associated with increases in endometrial thickness or increases in the rate of endometrial hyperplasia or carcinoma. Increases in venous thrombosis events were noted, which were not unexpected, since they were already well known to occur with estrogens and other SERMs.
Lasofoxifene
Lasofoxifene is another synthetic estrogen agonist–antagonist (SERM). It has a high affinity for both ERs, in a similar range to estradiol, and an order of magnitude higher than raloxifene, tamoxifen, or droloxifene. It has better oral bioavailability than several other SERMs.
Two doses of lasofoxifene 0.25 and 0.5 mg daily were evaluated in a large phase 3 double-blind, randomized trial, denoted as the PEARL (Postmenopausal Evaluation and Risk Reduction with Lasofoxifene) trial. A total of 8556 women were initially enrolled, and assessment of outcomes at 5 years was completed for 6614 (~77 %) of these [33]. There were significant increases in both lumbar spine and femoral neck BMD relative to placebo. There was a reduction in the risk of vertebral fractures by 31 % and 42 % with lasofoxifene given at 0.25 mg and 0.5 mg/day, respectively, while non-vertebral fractures were significantly reduced by 22 % with the 0.5 mg/day dose. The higher dose of lasofoxifene reduced the risk of total breast cancer by 79 % and of ER-positive invasive breast cancer by 83 % and the lower dose by 49 %.
Treatment with 0.5 mg of lasofoxifene per day was also associated with a reduction in major coronary heart disease events and stroke and was not associated with an increase in the risk of endometrial cancer or endometrial hyperplasia. However, reports of leg cramps, hot flushes, endometrial hypertrophy, uterine polyps, and vaginal candidiasis were significantly more common in women assigned to lasofoxifene than in those assigned to placebo.
Although lasofoxifene received approval for use in the European Union countries, it has never been marketed, despite the encouraging clinical results and fracture data compared with other SERMs.
Strontium
Strontium ranelate emerged in the 1990s as a potential treatment for osteoporosis. It was initially portrayed as an uncoupling agent, which stimulated bone formation while decreasing bone resorption. This was an attractive profile for a new drug for osteoporosis. The proposed dual mechanism of action even led to the introduction of the term “DABA” (dual-acting bone agent).
Two large pivotal phase 3 trials (Spinal Osteoporosis Therapeutic Intervention (SOTI) and Treatment of Peripheral Osteoporosis (TROPOS) ) were conducted within a total of 6740 Caucasian women with postmenopausal osteoporosis. In these trials, strontium ranelate showed efficacy in reducing fractures [34].
Strontium is the active component, while ranelate is an anion that enabled patent protection. Despite its apparent efficacy in reducing fractures, the mechanism by which this is achieved remains unclear [35]. The effects on bone resorption and formation are small, but there are substantial changes in BMD measurements largely due to substitution of strontium for calcium in bone mineral. It is known that strontium apatites have different solubility and physical characteristics than the calcium-containing hydroxyapatites and related minerals normally present in bone. Strontium salts have been used in the dental field as toothpaste additives for their protective effects on mineral dissolution. The simple classification of strontium as a dual-acting agent or an antiresorptive drug does not properly reflect these other properties on the physicochemical behavior of bone mineral, which may be an important part of its pharmacological actions.
Strontium ranelate has been quite widely used in osteoporosis treatment in several countries particularly in the elderly. Safety concerns about sensitivity reactions, venous thrombosis, and adverse cardiovascular effects are now limiting its use.
Calcitonins
Calcitonin is a peptide hormone secreted by the C-cells of the thyroid gland, which lowers blood calcium by inhibiting bone resorption. It acts directly on osteoclasts to produce a temporary cessation of their resorptive activity. Calcitonins were first used therapeutically in the 1970s for Paget’s disease of bone, in which they were moderately active. The potencies of calcitonins vary according to the species of origin, and porcine, human, and salmon calcitonins have all been used to varying extents in clinical practice. Salmon calcitonin emerged as the preferred of these, predominantly and curiously because it is more potent than human. It was eventually developed as a nasal inhalation rather than being given by injection. Studies with salmon calcitonin in osteoporosis only showed weak anti-fracture effects, and its use has been superseded by more effective treatments. Recent studies with oral calcitonin have also proved disappointing.
Calcitonin is still sometimes used for pain relief after vertebral fracture. The nasal forms of calcitonin have been recently withdrawn by the CHMP because of concerns about cancer risk.
Cathepsin K Protease Inhibitors
Cathepsin K (Cat K) is one of the primary enzymes involved in degrading type I collagen, the major component of the organic bone matrix . Cathepsin K is a member of the papain family of cysteine proteases and is highly expressed by activated osteoclasts but also in other cell types, giving rise to potential off-target effects (the skin, lungs, etc.). Cathepsins are lysosomal proteases that belong to the papain-like cysteine protease family. There are 11 different types (B, C, F, H, K, L, O, S, V, X, and W). There is incomplete homology of these enzymes among animal species, making animal experiments sometimes misleading. Making selective inhibitors is difficult but key to success. There has been intense activity in the pharmaceutical industry to create inhibitors as illustrated by the many patents that have been issued [36] (Fig. 2.4).
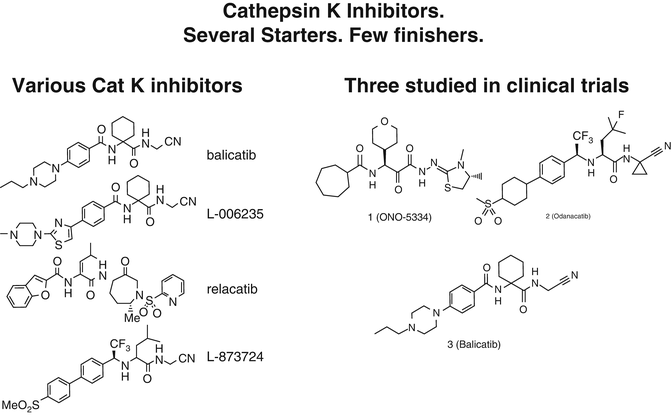
Fig. 2.4
Structures of various cathepsin K inhibitors. As discussed in the text, only odanacatib has been shown to reduce fractures in a phase 3 trial
Loss-of-function mutations in the cathepsin K gene lead to pycnodysostosis , a disorder characterized by osteosclerosis, bone fragility, and decreased bone turnover. This discovery led to a flurry of research activity especially in the mid-1990s. Cathepsin K therefore became an attractive therapeutic target in osteoporosis and also potentially in osteoarthritis and bone cancers. The concept is scientifically interesting and illustrates the process of modern drug design and development, based on identifying a target (cathepsin K) and then using medicinal chemistry to design and synthesize selective inhibitors [37]. Key components of a clinically viable inhibitor are oral bioavailability, high selectivity over related cathepsins, and a covalent, reversible warhead to bind to the active-site cysteine of the enzyme.
There have been many hurdles encountered during the discovery and development of cathepsin K inhibitors as drugs. These include species differences in amino acid sequences in the critical site of the target enzyme, species differences in bone metabolism, and also discrepancies between pharmacokinetic and pharmacodynamic (PK/PD) profiles due to unique tissue distribution of the inhibitor affecting both efficacy and side effects, originating from idiosyncratic intracellular or tissue distribution of some classes of compounds [38]. The antiresorptive properties of several cathepsin K inhibitors have been studied in phase I and phase II clinical trials. Phase III studies have recently been completed for odanacatib, the only cathepsin K inhibitor for which fracture reduction has been shown. Overall and despite the involvement of “big Pharma,” there have been more failures than successes in this field so far, with several “failures” in the development of Cat K inhibitors (e.g., balicatib, relacatib; see below).
Related posts:
 Epidemiology of Osteonecrosis of the Jaws from Antiresorptive Treatment
Fractures and Healing on Antiresorptive Therapy
Integrated Clinical View on Long-Term Management of Patients with Osteoporosis
Epidemiology of Osteonecrosis of the Jaws from Antiresorptive Treatment
Fractures and Healing on Antiresorptive Therapy
Integrated Clinical View on Long-Term Management of Patients with Osteoporosis
 Treatment Failure
Treatment Failure
 Anabolics
Anabolics
 Management of Osteonecrosis of the Jaw in Patients Receiving Antiresorptive Treatment
Management of Osteonecrosis of the Jaw in Patients Receiving Antiresorptive Treatment
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


