Pemetrexed is a pyrrolopyrimidine, multitargeted antifolate analog that targets multiple enzymes involved in folate metabolism, including thymidylate synthase (TS), dihydrofolate reductase (DHFR), glycinamide ribonucleotide (GAR) formyltransferase, and aminoimidazole carboxamide (AICAR) formyltransferase.1,2 This agent has broad-spectrum activity against solid tumors, including malignant mesothelioma and breast, pancreatic, head and neck, non–small-cell lung, colon, gastric, cervical, and bladder cancers.3–5
The third antifolate compound to have entered clinical practice is pralatrexate (10-propargyl-10-deazaaminopterin), a 10-deazaaminopterin antifolate that was rationally designed to bind with higher affinity to the reduced folate carrier (RFC)-1 transport protein, when compared with MTX, leading to enhanced membrane transport into tumor cells. It is also an improved substrate for the enzyme folylpolyglutamyl synthetase (FPGS), resulting in enhanced formation of cytotoxic polyglutamate metabolites.6,7 When compared with MTX, this analog is a more potent inhibitor of multiple enzymes involved in folate metabolism, including TS, DHFR, and GAR and AICAR formyltransferases. This agent is presently approved for the treatment of relapsed or refractory peripheral T-cell lymphomas.8
Mechanism of Action
The antifolate compounds are tight-binding inhibitors of DHFR, a key enzyme in folate metabolism.1 DHFR plays a pivotal role in maintaining the intracellular folate pools in their fully reduced form as tetrahydrofolates, and these compounds serve as one-carbon carriers required for the synthesis of thymidylate, purine nucleotides, and certain amino acids.
The cytotoxic effects of MTX, pemetrexed, and pralatrexate are mediated by their respective polyglutamate metabolites, with up to 5 to 7 glutamyl groups in a γ-peptide linkage. These polyglutamate metabolites exhibit prolonged intracellular half-lives, thereby allowing for prolonged drug action in tumor cells. Moreover, these polyglutamate metabolites are potent, direct inhibitors of several folate-dependent enzymes, including DHFR, TS, AICAR formyltransferase, and GAR formyltransferase.1
Mechanisms of Resistance
The development of cellular resistance to antifolates remains a major obstacle to its clinical efficacy.9,10 In experimental systems, resistance to antifolates arises from several mechanisms, including an alteration in antifolate transport because of either a defect in the reduced folate carrier or folate receptor systems, decreased capacity to polyglutamate the antifolate parent compound through either decreased expression of FPGS or increased expression of the catabolic enzyme γ-glutamyl hydrolase, and alterations in the target enzymes DHFR and/or TS through increased expression of wild-type protein or overexpression of a mutant protein with reduced binding affinity for the antifolate. Gene amplification is a common resistance mechanism observed in various experimental systems, including tumor samples from patients. In in vitro and in vivo experimental model systems, the levels of DHFR and/or TS protein acutely increase after exposure to MTX and other antifolate compounds. This acute induction of target protein in response to drug exposure is mediated, in part, by a translational regulatory mechanism, which may represent a clinically relevant mechanism for the acute development of cellular drug resistance.
Clinical Pharmacology
The oral bioavailability of MTX is saturable and erratic at doses greater than 25 mg/m2. MTX is completely absorbed from parenteral routes of administration, and peak serum levels are achieved within 30 to 60 minutes of administration.
The distribution of MTX into third-space fluid collections, such as pleural effusions and ascitic fluid, can substantially alter MTX pharmacokinetics. The slow release of accumulated MTX from these third spaces over time prolongs the terminal half-life of the drug, leading to potentially increased clinical toxicity. It is advisable to evacuate these fluid collections before treatment and monitor plasma drug concentrations closely.
Renal excretion is the main route of drug elimination, and this process is mediated by glomerular filtration and tubular secretion. About 80% to 90% of an administered dose is eliminated unchanged in the urine. Doses of MTX, therefore, should be reduced in proportion to reductions in creatinine clearance. Renal excretion of MTX is inhibited by probenecid, penicillins, cephalosporins, aspirin, and nonsteroidal anti-inflammatory drugs.
Pemetrexed enters the cell via the RFC system and, to a lesser extent, by the folate receptor protein. As with MTX, it undergoes polyglutamation within the cell to the pentaglutamate form, which is at least 60-fold more potent than the parent compound. This agent is mainly cleared by renal excretion, and in the setting of renal dysfunction, the terminal drug half-life is significantly prolonged to up to 20 hours. Pemetrexed, therefore, should be used with caution in patients with renal dysfunction. In addition, renal excretion is inhibited in the presence of other agents including probenecid, penicillins, cephalosporins, aspirin, and nonsteroidal anti-inflammatory drugs.
As with other antifolate analogs, pralatrexate is transported into the cell by the RFC carrier protein and then metabolized by FPGS to form longer chain polyglutamates, with up to four additional glutamate residues attached to the parent molecule. About 34% of the parent drug is cleared in the urine during the first 24 hours after drug administration. As such, caution is advised when using pralatrexate in patients with renal dysfunction. As with MTX and pemetrexed, the concomitant administration of other agents such as probenecid, penicillins, cephalosporins, aspirin, and nonsteroidal anti-inflammatory drugs, may inhibit renal clearance.
Toxicity
The main side effects of MTX are myelosuppression and gastrointestinal (GI) toxicity, which are usually completely reversed within 14 days, unless drug-elimination mechanisms are impaired. In patients with compromised renal function, even small doses of MTX may result in serious toxicity. MTX-induced nephrotoxicity is thought to result from the intratubular precipitation of MTX and its metabolites in acidic urine. Antifolates may also exert a direct toxic effect on the renal tubules. Vigorous hydration and urinary alkalinization have greatly reduced the incidence of renal failure in patients on high-dose regimens. Acute elevations in hepatic enzyme levels and hyperbilirubinemia are often observed during high-dose therapy, but these levels usually return to normal within 10 days. Methotrexate given concomitantly with radiotherapy may increase the risk of soft tissue necrosis and osteonecrosis.
The original rationale for high-dose MTX therapy was based on the concept of selective rescue of normal tissues by the reduced folate leucovorin (LV). However, recent data suggest that high-dose MTX may also overcome resistance mechanisms caused by impaired active transport, decreased affinity of DHFR for MTX, increased levels of DHFR resulting from gene amplification, and/or decreased polyglutamation of MTX.
The main toxicities of pemetrexed and pralatrexate include dose-limiting myelosuppression, mucositis, and skin rash, usually in the form of the hand-foot syndrome (HFS). Other toxicities include reversible transaminasemia, anorexia and fatigue syndrome, and GI toxicity. These side effects are reduced by supplementation with folic acid (350 μg orally daily) and vitamin B12 (1,000 mg subcutaneously given at least 1 week before starting therapy, and then repeated every three cycles). To date, there is no evidence to suggest that vitamin supplementation adversely affects the clinical efficacy of pemetrexed or pralatrexate.
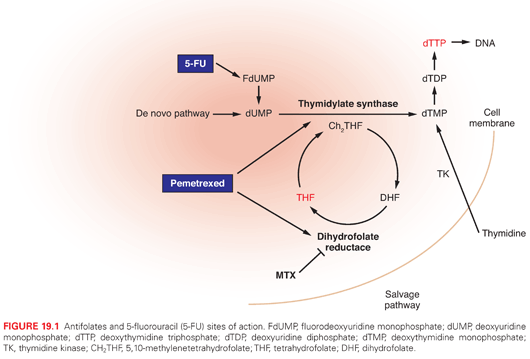
The fluoropyrimidine, 5-fluorouracil (5-FU) was synthesized by Charles Heidelberger in the mid 1950s. Uracil is a normal component of RNA; as such, the rationale leading to the development of the drug was that cancer cells might be more sensitive to decoy molecules that mimic the natural compound than normal cells. 5-FU and its derivatives are an integral part of treatment for a broad range of solid tumors (see Table 19.1), including GI malignancies (esophageal, gastric, pancreatic, colorectal, anal, and hepatocellular cancers), breast, head and neck, and skin cancers.11 It continues to serve as the main backbone for combination regimens used to treat metastatic colorectal cancer (mCRC) and as adjuvant therapy of early-stage colon cancer.
Mechanism of Action
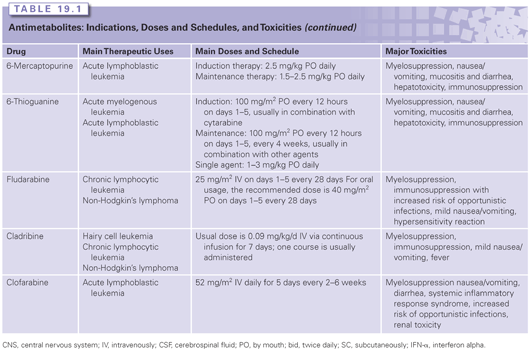
5-FU enters cells via the facilitated uracil base transport mechanism and is then anabolized to various cytotoxic nucleotide forms by several biochemical pathways. It is thought that 5-FU exerts its cytotoxic effects through various mechanisms, including (1) the inhibition of TS, (2) incorporation into RNA, and (3) incorporation into DNA (Fig. 19.1). In addition to these mechanisms, the genotoxic stress resulting from TS inhibition may also activate programmed cell-death pathways in susceptible cells, which leads to the induction of parental DNA fragmentation.
Mechanisms of Resistance
Several resistance mechanisms to 5-FU have been identified in experimental and clinical settings. Alterations in the target enzyme TS represent the most commonly described mechanism of resistance. In vitro, in vivo, and clinical studies have documented a strong correlation between the levels of TS enzyme activity/TS protein and chemosensitivity to 5-FU. In this regard, cell lines and tumors with higher levels of TS are relatively more resistant to 5-FU. Mutations in the TS protein have been identified that lead to reduced binding affinity of the 5-FU metabolite fluorodeoxyuridine monophosphate (FdUMP) to the TS protein. Reduced expression and/or diminished activity of key activating enzymes may interfere with the formation of cytotoxic 5-FU metabolites. Decreased expression of mismatch repair enzymes, such as human mutL homolog 1 (hMLH1) and human mutS homolog 2 (hMSH2), and increased expression of the catabolic enzyme dihydropyrimidine dehydrogenase (DPD) are associated with fluoropyrimidine resistance. At this time, the relative contribution of each of these mechanisms in the development of cellular resistance to 5-FU in the actual clinical setting remains unclear.
Clinical Pharmacology
5-FU is not orally administered, given its erratic bioavailability resulting from high levels of the catabolic enzyme DPD present in the gut mucosa. After intravenous bolus doses, metabolic elimination is rapid, with a half-life of 8 to 14 minutes. More than 85% of an administered dose of 5-FU is enzymatically inactivated by DPD, the rate-limiting enzyme in the catabolism of 5-FU.
A pharmacogenetic syndrome has been identified in which partial or compete deficiency in the DPD enzyme is present in 3% to 5% and 0.1% of the general population, respectively. As DPD catalyzes the rate-limiting step in the catabolic pathway of 5-FU, a deficiency of DPD can result in a clinically dangerous increase in the anabolic products of 5-FU. Unfortunately, patients with DPD deficiency do not manifest a phenotype only until they are treated with 5-FU, and in that setting, they can develop severe GI toxicity in the form of mucositis and/or diarrhea, myelosuppression, neurologic toxicity, and in rare cases, death. In patients being treated with 5-FU or any other fluoropyrimidine, it is important to consider DPD deficiency in patients who present with excessive, severe toxicity.12 It is now increasingly appreciated that DPD mutations are unable to account for all of the observed cases of excessive 5-FU toxicity, because up to 50% of patients who experience 5-FU toxicity will have no documented alterations in the DPD gene. Moreover, individuals with normal DPD enzyme activity may be diagnosed with high plasma levels of 5-FU, resulting in increased toxicity. Although DPD enzyme activity can be assayed from peripheral blood mononuclear cells in a specialized laboratory, routine phenotypic and genotypic screenings for DPD deficiency prior to 5-FU therapy are not yet available.
Biomodulation of 5-FU
Significant efforts have focused on enhancing the antitumor activity of 5-FU through biochemical modulation in which 5-FU is combined with various agents, including leucovorin, MTX, N-phosphonacetyl-l-aspartic acid, interferon-α, interferon-γ, and a whole host of other agents.13 For the past 20 to 25 years, the reduced folate LV has been the main biochemical modulator of 5-FU. An alternative approach has been to alter the schedule of 5-FU administration. Given the S-phase specificity of this agent, prolonged exposure of tumor cells to 5-FU would increase the fraction of cells being exposed to the drug. Overall response rates are significantly higher in patients treated with infusional schedules of 5-FU than in those treated with bolus 5-FU, and this improvement in response rate has translated into an improved progression-free survival. Moreover, the overall safety profile is improved with infusional regimens. A hybrid schedule of bolus and infusional 5-FU was originally developed in France, and this regimen has shown superior clinical activity compared with bolus 5-FU schedules. This hybrid schedule has now been simplified by using only the 46-hour infusion of 5-FU and completely eliminating the 5-FU bolus doses.
Toxicity
The spectrum of 5-FU toxicity is dose- and schedule-dependent (Table 19.2). The main side effects are diarrhea, mucositis, and myelosuppression. The dermatologic HFS is more commonly observed with infusional 5-FU therapy. Acute neurologic symptoms have also been reported, and they include somnolence, cerebellar ataxia, and upper motor signs. Treatment with 5-FU can, on rare occasions, cause coronary vasospasm, resulting in a syndrome of chest pain, cardiac enzyme elevations, and electrocardiographic changes. Cardiac toxicity seems to be related more to infusional 5-FU than bolus administration.14
Capecitabine is an oral fluoropyrimidine carbamate that was rationally designed to allow for selective 5-FU activation in tumor tissue.15 This oral agent was initially approved in anthracycline-and taxane-resistant breast cancer and subsequently approved for use in combination with docetaxel as second-line therapy in metastatic breast cancer and in combination with lapatinib, a tyrosine-kinase inhibitor of human epidermal growth factor receptor type 2 (HER2) and epidermal growth factor receptor (EGFR) in women with HER2-positive metastatic breast cancer following progression on trastuzumab-based therapy.16 This agent is also approved by the U.S. Food and Drug Administration (FDA) for the first-line treatment of mCRC and as adjuvant therapy for stage III colon cancer when fluoropyrimidine therapy alone is preferred.17 In Europe and throughout much of the world, the combination of capecitabine plus oxaliplatin (XELOX) is approved for the treatment of mCRC as well as for the adjuvant therapy of stage III colon cancer.18 In addition, recent studies have documented the noninferiority of capecitabine to 5-FU when combined with cisplatin in the treatment of metastatic gastric cancer.
Clinical Pharmacology
Capecitabine is rapidly and extensively absorbed by the gut mucosa, with nearly 80% oral bioavailability. It is inactive in its parent form and undergoes enzymatic conversion via three successive steps. Of note, the third and final step occurs in tumor tissue and involves the conversion of 5′-deoxy-5-fluorouridine to 5-FU by the enzyme thymidine phosphorylase (TP), which is expressed at much higher levels in tumors when compared with corresponding normal tissue. Capecitabine and capecitabine metabolites are primarily excreted by the kidneys, and in contrast to 5-FU, caution must be taken in the presence of renal dysfunction, with appropriate dose modification. The use of capecitabine is absolutely contraindicated in patients whose creatinine clearance is less than 30 mL per minute. The FDA and Roche have added a black box warning and strengthened the precautions section on the capecitabine label about the drug–drug interaction between warfarin and capecitabine-based chemotherapy. It is generally recommended to do weekly monitoring of the coagulation parameters (prothrombin time/international normalized ratio [PT/INR]) for all patients receiving concomitant warfarin and capecitabine, with an appropriate adjustment of warfarin dose.
Toxicity
Similar to what is observed with infusional 5-FU, the main side effects of capecitabine include diarrhea and HFS. Of note, the incidence of myelosuppression, neutropenic fever, mucositis, alopecia, and nausea/vomiting is lower with capecitabine when compared with 5-FU. Elevations in indirect serum bilirubin can be observed, but are usually transient and clinically asymptomatic. Patients in the United States appear to be unable to tolerate as high doses of capecitabine as European patients, either as monotherapy or in combination with other cytotoxic chemotherapy.19 Although the underlying reasons for this discrepancy are not known, it may in part be related to the increased fortification of the US diet with folate and the increased focus on vitamin and folic acid supplementation.
S-1
S-1 is an oral fluoropyrimidine that consists of tegafur (FT), a prodrug of 5-FU, combined with two 5-FU biochemical modulators: 5-chloro-2,4-dihydroxypyridine (gimeracil or CDHP), a competitive inhibitor of DPD, and oteracil potassium, which inhibits phosphorylation of 5-flurouracil in the GI tract, thereby decreasing serious GI toxicities such as nausea/vomiting, mucositis, and diarrhea.20 As with other oral agents, S-1 offers several advantages over 5-FU, including ease of administration, no risks associated with use of central venous access such as infection, thrombosis, etc., and reduced toxicities, especially neurotoxicity. Although S-1 has yet to be approved by the FDA, it has been approved for the treatment of gastric cancer, head and neck, colorectal cancer (CRC), non–small-cell lung, breast, pancreatic, and biliary tract cancers in several countries in Asia and for the treatment of advanced gastric cancer in combination with cisplatin in a large number of European countries.
Clinical Pharmacology
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


