Antigen Processing and Presentation
Ted H. Hansen
Paul A. Roche
ANTIGEN PRESENTATION PATHWAYS
As part of the adaptive immune response, T cells mount immune responses to diseased cells and abnormal cells. Remarkably, the mechanism by which T cells discriminate self from non-self is based on pathogen- or tumor-derived proteins displayed on the cell surface by self-major histocompatibility complex (MHC) molecules. More specifically, when T cells detect diseased cells displaying MHC molecules loaded with peptides derived from foreign peptides, effector mechanisms are initiated to eliminate the diseased cell. By contrast, T cells that detect cells displaying MHC molecules loaded with peptides derived from self-proteins are either eliminated during ontogeny by negative selection or suppressed by peripheral tolerance mechanisms. Thus, at the molecular level, the T-cell receptors on T cells discriminates MHC loaded with self- versus non-self-peptides displayed at the cell surface of normal and diseased cells. The mechanisms by which peptide ligands for MHC molecules are generated is referred to as antigen processing, whereas the mechanism by which peptide/MHC complexes are displayed at the cell surface is called antigen presentation.
To assure the appropriate effector mechanism is generated to detect cells infected with pathogens in the cytosol or endocytic compartments, the antigen presentation pathways for classical cluster of differentiation (CD)8+ versus CD4+ T cells are different. More specifically, antigen derived from intracellular pathogens is typically bound to MHC-I proteins that are uniquely detected by CD8 T cells, whereas antigen derived from extracellular pathogens is typically bound to MHC-II proteins that are uniquely detected by CD4 T cells. To preferentially bind peptides derived from proteins synthesized inside the cell, the processing of peptides that bind MHC-I proteins occurs in the cytosol and the loading of these peptides occurs in the endoplasmic reticulum (ER). This pathway is commonly referred to as the classical MHC-I antigen-binding pathway. By contrast, to preferentially bind peptides from proteins synthesized outside the cell, the processing and loading of antigenic peptide ligands for MHC-II proteins occurs in an endocytic pathway terminating in the lysosomes. This pathway is typically referred to the classical MHC-II antigen-binding pathway. The differences between the MHC-I and MHC-II antigen presentation pathways are determined by their differential interaction with molecular chaperones. These same molecular chaperones enforce the quality control of antigen presentation to assure appropriate T cells are activated for detection and elimination of pathogen-infected cells or tumors.
ANTIGEN PROCESSING AND PRESENTATION BY MAJOR HISTOCOMPATIBILITY CLASS I (MHC-I) PROTEINS
Origin of MHC-I binding peptides
As detailed in Chapter 21, MHC-I proteins preferentially bind peptides of 8 to 10 amino acid lengths, and which peptides bind is determined by MHC-I allele-specific polymorphic residues that form the architecture of its peptide binding groove. Each MHC-I allele typically binds peptides with a consensus binding motif requiring a relatively specific amino acid central anchor and a C-terminal hydrophobic amino acid. Given the lack of restraints in most peptide positions and the large number of proteins synthesized by each cell relative to the number of MHC molecules, the selective processes controlling which peptides get presented is of considerable importance for the detection of pathogens and malignancies.
The source of the peptides that get presented during infection has been a question of considerable investigation. It has been known for some time that MHC-I-binding peptides are derived from membrane-bound, secreted, cytosolic, and nuclear proteins that are almost exclusively generated by the proteasome in the cytosol. However, the uncertainty of peptide source is based on two apparently incongruent observations. Most proteins from which MHC-I-binding peptides are derived have slow rates of turnover in cells ranging from hours to days; viral-derived peptides are presented by infected cells to CD8 T cells within a few minutes. This disparity between the degradation versus presentation kinetics is potentially explained by cumulative observations that a fraction of newly synthesized polypeptides are rapidly degraded, providing a peptide reservoir sufficient to support a robust CD8 T-cell response. Supporting evidence for this conclusion comes from studies that block protein synthesis with cycloheximide and detected a rapid decrease in the peptide supply for MHC-I binding.1,2 One of the more elegant ways to monitor peptide transport into the ER was by transporter associated with antigen presentation (TAP) mobility in the ER membrane by photobleaching; establishing TAP mobility was proportional to the peptides in the cytosol. This assay was used to show a 90% reduction in peptide supply after 30 m treatment with cycloheximide, thus kinetically linking presentation closely to translation.3 Indeed, there are now several reports that within 30 minutes after synthesis, antigenic peptides are presented at
the cell surface at levels that activate T cells.4,5 What remained unclear from these studies is the biochemical and mechanistic basis for the generation of these rapidly transcribed proteins from which MHC-I-binding peptides are derived.
the cell surface at levels that activate T cells.4,5 What remained unclear from these studies is the biochemical and mechanistic basis for the generation of these rapidly transcribed proteins from which MHC-I-binding peptides are derived.
In a model that has gained wide acceptance, Yewdell and colleagues proposed that a rapid supply of MHC-I ligands is derived from aberrant protein production they termed defective ribosomal products or DriPs.6,7 In their model, it was speculated that DriPs could be derived from unfolded or misfolded proteins of the proper sequence and length, proteins with errors in sequence or posttranslational modifications, prematurely terminated proteins, and proteins translated from the wrong start codon.8 Regarding aberrant translations, there is evidence that MHC-presented peptides can arise from alternative reading frames, generating what has been called cryptic epitopes.9 For example, Schwab et al. showed that inhibition of the eukaryotic translational initiation factor eIF2α resulted in the synthesis of cryptic peptides and intriguingly, viruses inhibit host translation by inactivating eIF2α.10,11 However, most MHC-I-presented peptides have native amino acid sequence and are derived from standard messenger ribonucleic acid (mRNA) translation products, suggesting cryptic epitopes represent only a minor component of MHC-presented peptides. In addition, the contribution of peptides derived from misfolded proteins in the secretory pathway is also likely modest. Although antigenic peptides from misfolded tyrosinase were found to be preferentially presented,12 other studies show that several mutations causing misfolding do not lead to enhanced antigen presentation. Furthermore, ER quality control is typically too slow to account for the rapid supply of MHC-I-binding peptides.13 The reason for this kinetic delay is that substrates for ER-associated degradation (ERAD) must be 1) translocated into the ER, 2) detected presumably by ER chaperones as being misfolded, 3) ubiquitinated and extracted from the ER, and 4) degraded by the proteasome in the cytosol. Thus the predominance of MHC-I-presented peptides likely does not come from cryptic epitopes or misfolded ER proteins identified by ER quality control.
To obviate trafficking into the ER, misfolded secretory proteins could be degraded rapidly based on degron detection such as unshielded hydrophobicity.14 Alternatively, early in translation a lack of signal recognition particle engagement could lead to the mistargeting of secretory proteins to the cytosol. In support of this model, Schlosser et al.15 showed that an epitope encompassing the signal peptidase cleavage site was efficiently presented by MHC-I proteins. To rapidly generate peptides, it has been speculated that cells may have select ribosomes called “immunoribosomes.”16 Such immunoribosomes might be more adept at generating peptides with TAP access perhaps by compartmentalization and/or targeting proteins to the 20S proteasome for immediate destruction. In any case, there is considerable evidence that antigenic peptides can be rapidly presented by MHC-I within minutes after translation of the protein source. The physiologic significance of this rapid kinetics of antigen presentation is that it allows CD8 T cells to kill virus-infected cells before viral replication is completed and progeny are released. However, whether the production of peptides bound to MHC class I molecules is a deliberate process for degrading defective translation products and/or a stochastic event of normal protein translation remains to be determined.17
Peptide Trimming in the Cytosol
Degradation of cellular and antigenic peptides bound to MHC-I is largely mediated in the cytosol by the proteasome. The proteasome is responsible for the degradation of the majority of cytosolic and nuclear proteins, and in most cases proteasome targeted proteins are ubiquitinated. Ubiquitin (Ub) is typically coupled to internal lysine residues of proteins substrates, but coupling can also occur at the N terminus or on internal cysteine, serine, or threonine residues.18,19,20,21 In this orchestrated process, activated Ub is transferred from one predominant Ub-activating enzyme (E1) to one of 30 to 40 mammalian Ub conjugating enzymes (E2s) and then to the substrate that is bound by one of hundreds of different ubiquitin protein ligases (E3s). The E3s are the major determinant of substrate specificity. This process can be repeated to form polyUb chains whereupon the next Ub moiety is added to one of the seven internal lysine residues of Ub. Proteins coupled with polyUb chains of four or more Ub moieties linked through Lys48 Ub residues are the prototypic signal for proteasome mediated degradation.
The cylindrical 20S catalytic particle (CP) of the proteasome is formed by four stacked rings of seven subunits each. The inner two rings of the 20S proteasome are assembled from the beta subunits, three of which have catalytic activity with chymotryptic, tryptic, or caspase activity. Thus each proteasome has six active sites with the ability to cleave after most types of peptide bonds, although to differing efficiencies based on flanking residues. The catalytic sites are exposed to the interior of the central chamber of the 20S cylinder. The two outer rings of the 20S proteasome are assembled from alpha subunits forming a pore of 13 Angstrom, mandating protein substrates be partially denatured prior to entering the interior of the chamber. The gate of the 20S CP is normally closed, and access to the interior of the 20S proteasome is controlled by the complex of proteins termed the 19S regulatory particle that binds to each end of the 20S cylinder. The full assemblage of 19S-20S-19S complex constitutes the 26S proteasome. The lid of the 19S regulator particle binds and deubiquitinates protein substrates before they gain entry in the 20S CP; the base of the 19S regulatory particle contains ATPases that promote substrate unfolding and open the gate of the 20S CP to provide access of denatured protein substrates to the catalytic activity of the inner chamber.
The evolutionarily conserved function of the proteasome is for recycling amino acids and ubiquitin moieties. However, in mammals, modifications to the proteasome have been made to promote antigen presentation. In response to interferon (IFN)γ stimulation that occurs during inflammation, three catalytic subunits of the constitutively expressed proteasome are replaced by IFNγ-induced homologs forming what is called the immunoproteasome. More specifically, subunits β1, β2, and β5 of the constitutive proteasome are replaced by subunits β1i (LMP2), β2i (MECL-1), and β5i (LMP7) of the immunoproteasome. Suggesting an immunologic function,
region of the mouse and human MHCs juxtaposed to genes encoding the TAP heterodimer. Consistent with their immunologic relevance, the immunoproteasomes display enhanced cleavage of protein substrates after hydrophobic and basic amino acids, thus generating peptides with C-terminal residues preferred for binding to many MHC-I alleles. IFNγ stimulation also induces the expression of PA28α and PA28β that form the 11S regulatory particle, which can also bind to either end of the 20S CP. Interestingly, the PA28 regulatory particle does not contain ATPases, but it does induce conformational changes to open the 20S CP gate. Thus, PA28 may function to prevent the overdigestion of short peptides. This is an important issue, as it has been estimated that a nonamer peptide in the cytosol has a half-life of only 7 seconds.22 However, studies testing PA28α/β-deficient mice demonstrated that PA28α/β is not required for overall antigen presentation during virus infection, but may affect degradation of select substrates.23 In any case, the inclusion of the IFNγ-inducible β subunits into the 20S CP of immunoproteasome enhances antigen presentation by generating peptides with preferred C-terminal anchor residues, and in some same cases inclusion of the PA28 regulatory particle may prevent overdegradation of immunologically relevant proteins.
region of the mouse and human MHCs juxtaposed to genes encoding the TAP heterodimer. Consistent with their immunologic relevance, the immunoproteasomes display enhanced cleavage of protein substrates after hydrophobic and basic amino acids, thus generating peptides with C-terminal residues preferred for binding to many MHC-I alleles. IFNγ stimulation also induces the expression of PA28α and PA28β that form the 11S regulatory particle, which can also bind to either end of the 20S CP. Interestingly, the PA28 regulatory particle does not contain ATPases, but it does induce conformational changes to open the 20S CP gate. Thus, PA28 may function to prevent the overdigestion of short peptides. This is an important issue, as it has been estimated that a nonamer peptide in the cytosol has a half-life of only 7 seconds.22 However, studies testing PA28α/β-deficient mice demonstrated that PA28α/β is not required for overall antigen presentation during virus infection, but may affect degradation of select substrates.23 In any case, the inclusion of the IFNγ-inducible β subunits into the 20S CP of immunoproteasome enhances antigen presentation by generating peptides with preferred C-terminal anchor residues, and in some same cases inclusion of the PA28 regulatory particle may prevent overdegradation of immunologically relevant proteins.
The proteasome generates the final C-terminal cleavage of most MHC-I-binding peptides. A recently reported exception to this is the generation of the tumor antigen MAGE-A3 by insulin-degrading enzyme.23 Whereas insulin degrading enzyme is solely responsible for the generation of the human leukocyte antigen (HLA)-A1-restricted MAGE-A3 epitope, the HLA-B40-restricted MAGE-A3 epitope was proteasome dependent. Although there are several N-terminal proteases in the cytosol, they have only a limited role in generation of the MHC-I-binding peptides. More specifically, tripeptidyl peptidase II, leucine amino peptidase, bleomycin hydrolase, and puromycin-sensitive aminopeptidase have all been found to affect generation of select MHC-I-binding epitopes, but these proteases have an overall negative effect on antigen presentation by MHC-I. Interestingly, the metalloprotease nardilysin that cleaves substrates on the N-terminus of arginine residues in dibasic pairs was implicated in three cytotoxic T lymphocyte epitopes and may be more generalizable given the preference for basic N-terminal resides for MHC-I binding.24 However, as noted in the following, significant N terminal trimming of MHC-I-binding peptide occurs after transport from the cytosol into the ER.
Peptide Transport into the Endoplasmic Reticulum
Landmark studies of the mutagenized and immunoselected mouse cell line RMA-S resulted in the discovery of how peptides are transported from the cytosol into the ER lumen.25,26,27 The low level of surface expression of MHC-I molecules by RMA-S that could be rescued by either low temperatures or culturing with known MHC-I-binding peptides was attributed to lack of peptide transport by TAP. TAP is a member of the adenosine triphosphate (ATP) binding cassette family. Mechanistically, hydrolysis of ATP is required both for peptide binding to the cytosolic face of TAP as well as peptide transport into the ER. Structurally, TAP is a heterodimeric complex of TAP1 and TAP2 subunits both encoded within the central region of the MHC of mouse and human. The translocation pore of TAP is formed by six transmembrane domains of each subunit, whereas the remaining transmembrane domains (four for TAP1 and three for TAP2) are involved in interaction with peptide loading complex (PLC).28 Awaiting transport, both the N- and C-termini of peptides are bound to TAP29; peptides with higher affinity for binding TAP have a greater likelihood of transport, MHC binding, and presentation to T cells during infection.30,31 TAP typically transports peptides of 8 to 16 residues, although longer peptides are less efficiently transported. As discussed in the next section, peptides with N-terminal extensions are trimmed in the ER.
Peptide Trimming in the Endoplasmic Reticulum
As noted previously, TAP transports many peptides with C-terminal hydrophobic residues required for MHC binding, but with N-terminal extensions, which need to be cleaved to conform to the MHC-binding motif of the expressed alleles. It is now clear that N-terminal trimming within the ER is carried out by the ER-associated aminopeptidase (ERAAP; ERAP1 in mice and ERAP1 in humans). Interestingly, unlike other aminopeptidases, ERAP1 preferentially cleaved peptides of 9 to 16 amino acids thus matching the same peptide length preference as transported by TAP.32 More specifically, ERAP1 was found to degrade a model 13-mer to a 9-mer and then stop, an apparent adaptation to maximize optimal MHC-I binding. The length preference of ERAP1 could be explained by extended peptides binding to MHC-I before N-terminal trimming. However, this model is not supported by structural analyses of ERAP1 and the location of its active site.33 Alternatively, recent analyses support a “molecular ruler” model. In this model, a hydrophobic pocket of ERAP1 binds the C-terminal peptide residue thereby positioning the peptide so that N-terminal cleavage occurs about nine amino acids away.32,33 Thus, ERAAP/ERAP1 has unique structural features explaining its ability to trim extended peptide precursors while sparing ones of optimal length for MHC-I binding. As further evidence of their specialized functions in antigen presentation by MHC-I proteins, ERAP1 preferentially binds and processes peptides with hydrophobic C-terminal residues consistent with the majority of the TAP-transported MHC-I-binding peptides. Interestingly, humans have a second ER-associated peptidase with homology to ERAP1 designated ERAP2.34 ERAP2 preferentially binds peptides with C-terminal basic residues and thus may function in human cells to provide peptides for HLA-A3, -Aw68, and -B27 alleles that bind such peptides. Additionally, in humans the combined activity of both ERAP1 and ERAP2 (most likely as heterdimers) is required for the presentation of certain epitopes.35 By contrast, in mice ERAAP/ERAP1 is sufficient to service their MHC-I alleles, all of which bind peptides with hydrophobic C-termini. As additional evidence for their tailoring for MHC-I-antigen presentation, the expression and trimming activity of ERAP1 and ERAP2 are upregulated upon IFNγ stimulation.
The impact of ERAP on specific epitopes and the overall MHC-binding peptide repertoire has been analyzed using
cell lines36,37 and ERAAP-knockout mice.38,39 These studies have shown that cytotoxic T lymphocyte detection of some but not all MHC-I/peptide complexes are ERAP1-dependent. For example, in vitro ribonucleic acid interference experiments suggested that ERAP1 is involved in formation of onethird of peptide/MHC-I complexes.36,37 ERAAP-deficient mice had a 20% lower expression of Kb, Db, Kd, and Dd alleles at the cell surface but a 70% lower expression of Ld. Of these mouse alleles, only Ld preferentially binds peptides with the motif of Pro in the second position, although about 20% of human HLA alleles also prefer peptides with a Pro in the second position. Notably, peptides with Pro in the second position are only transported by TAP with N-terminal extensions and are thus dependent upon ERAP1 for their generation. In vivo relevance of this conclusion was demonstrated by the observation that ERAAP-deficient BALB/c mice were found to be susceptible to Toxoplasma gondii infection resulting from the fact that the immunodominant epitope presented to CD8 T cells is Ld restricted.40 Interestingly the ERAAP-deficient mice were also found to present several unique peptides that elicited potent CD8 T-cell responses demonstrating ERAAP functions, as a peptide editor altering the repertoire of peptides presented.38
cell lines36,37 and ERAAP-knockout mice.38,39 These studies have shown that cytotoxic T lymphocyte detection of some but not all MHC-I/peptide complexes are ERAP1-dependent. For example, in vitro ribonucleic acid interference experiments suggested that ERAP1 is involved in formation of onethird of peptide/MHC-I complexes.36,37 ERAAP-deficient mice had a 20% lower expression of Kb, Db, Kd, and Dd alleles at the cell surface but a 70% lower expression of Ld. Of these mouse alleles, only Ld preferentially binds peptides with the motif of Pro in the second position, although about 20% of human HLA alleles also prefer peptides with a Pro in the second position. Notably, peptides with Pro in the second position are only transported by TAP with N-terminal extensions and are thus dependent upon ERAP1 for their generation. In vivo relevance of this conclusion was demonstrated by the observation that ERAAP-deficient BALB/c mice were found to be susceptible to Toxoplasma gondii infection resulting from the fact that the immunodominant epitope presented to CD8 T cells is Ld restricted.40 Interestingly the ERAAP-deficient mice were also found to present several unique peptides that elicited potent CD8 T-cell responses demonstrating ERAAP functions, as a peptide editor altering the repertoire of peptides presented.38
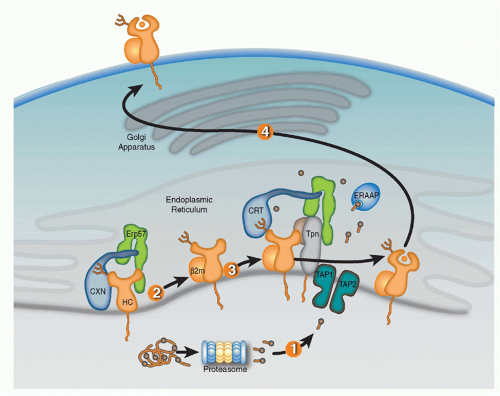 FIG. 22.1. Schematic Representation of the Sequential Events of Antigen Processing and Presentation by Major Histocompatibility Complex (MHC)-I Proteins. 1: Peptides are processed by the proteasome and translocated into the ER by TAP1/TAP2 heterodimers. 2: MHC-I heavy chains (HCs) awaiting assembly with β2m are bound to calnexin (CXN)/ERp57 complexes. 3: After assembly with β2m, HC/β2m heterodimers enter the peptide loading complex wherein tapasin (Tpn) bridges HC with transporter associated with antigen presentation (TAP)/TAP2, and calreticulin (CRT)/ERp57 complexes bridge HC with Tpn. 4: Following the binding of a suitable peptide, which may require final N-terminal trimming by ERAAP, fully assembled MHC-I proteins (HC/β2m/peptide) traffic to the plasma membrane via the secretory pathway. |
Chaperone-assisted Peptide Loading in the Endoplasmic Reticulum
Full assembly of the MHC-I heavy chains (HCs) with β2m and peptide within the ER is orchestrated by molecular chaperones that keep folding intermediates in a conformation capable of attaining full assembly (Fig. 22.1). Nascent HCs are transiently associated with the membrane-anchored
lectin calnexin (CXN) that, like its soluble paralog calreticulin (CRT), functions as a general chaperone for assembly of oligomeric glycoproteins. Quality control of glycoprotein assembly by CXN and CRT is carried out by monitoring terminal glucose residues on their N-linked glycans. More specifically, the folding sensor UDP-glucose glycoprotein glucotransferase allows incompletely assembled substrates to reassociate with CXN or CRT, and cycles of glucose addition and removal continue until substrates are either correctly folded and assembled or targeted for ERAD. After the MHC-I HC assembles with β2m, the HC/βm heterodimer exchanges CXN for CRT and enters the PLC consisting of CRT, ERp57, tapasin (Tpn), and TAP.41,42,43,44 Optimal antigen presentation by MHC-I is dependent upon all four components of the PLC (Tpn, TAP, CRT, and ERp57) that function to improve peptide loading in MHC-I proteins and their surface display.45,46
lectin calnexin (CXN) that, like its soluble paralog calreticulin (CRT), functions as a general chaperone for assembly of oligomeric glycoproteins. Quality control of glycoprotein assembly by CXN and CRT is carried out by monitoring terminal glucose residues on their N-linked glycans. More specifically, the folding sensor UDP-glucose glycoprotein glucotransferase allows incompletely assembled substrates to reassociate with CXN or CRT, and cycles of glucose addition and removal continue until substrates are either correctly folded and assembled or targeted for ERAD. After the MHC-I HC assembles with β2m, the HC/βm heterodimer exchanges CXN for CRT and enters the PLC consisting of CRT, ERp57, tapasin (Tpn), and TAP.41,42,43,44 Optimal antigen presentation by MHC-I is dependent upon all four components of the PLC (Tpn, TAP, CRT, and ERp57) that function to improve peptide loading in MHC-I proteins and their surface display.45,46
Knockout cell lines, genetically deficient mouse strains, and mutagenesis studies have provided key insights in the selective roles of PLC components in MHC-I assembly. CRT association with MHC-I proteins is dependent upon the N-linked glycan at residue Asn86 of the HC. Interestingly, the location of the glycan is important for CRT but not CXN association with HCs, likely reflecting geometric constraints imposed by PLC assembly. CRT-deficient cells have suboptimal peptide loading resulting in reduced surface MHC-I expression.47 ERp57 is a thiol oxidoreductase that mediates disulfide bond formation of different substrates and is commonly associated with CXN and CRT. Interestingly, however, the function of ERp57 as a component of the PLC appears to be independent of the MHC-I redox state. ERp57 forms a disulfide bond with Tpn that is required for PLC construction.48,49 It was originally proposed that the ERp57-Tpn complex prevents reduction of the α2 disulfide HC bond, thereby keeping HC in a peptide-receptive state.50 However, other studies have detected little evidence the ERp57 controls redox state of MHC-I and more recently, the crystal structure of the ERp57-Tpn complex suggests that the role of the ERp57 in the PLC is structural, facilitating recruitment of peptide-accessible MHC-I.49,51,52 As a likely reflection of its fundamental role in PLC construction, ERp57-deficient cells were found to have impaired peptide loading, surface expression, and antigen presentation by MHC-I proteins.
Tpn, an MHC-I-dedicated molecular chaperone, is required to bridge β2m-assembled HCs with TAP.53 Playing a redundant role with other members of the PLC, Tpn also functions in ER retention of MHC with suboptimal peptide cargo.54,55 Perhaps most importantly, Tpn functions as a peptide editor by stabilizing peptide-accessible MHC-I proteins and optimizing peptide cargo before release from the PLC. The mechanism of peptide editing by Tpn of MHC-I proteins was revealed using recombinant Tpn tethered to HLA,56 and using recombinant Tpn-ERp57 conjugates added to Tpn-deficient cells.57 These and other reports support the model that Tpn promotes the peptide exchange of MHC molecules in an affinity-dependent manner. The consensus model is that Tpn stabilizes the peptide binding groove of MHC-I in an “open” peptide-accessible conformation. The association of Tpn with peptide-accessible MHC-I then promotes peptide exchange until a peptide of suitable affinity binds to complete the folding of the ligand binding groove. This peptide-induced folding then induces the release of fully assembled MHC-I from the PLC. Logistically, how peptide editing by Tpn might occur was provided by mutagenesis studies of the MHC-I/Tpn interaction site. Two sites on the HC are critical for Tpn interaction: one site is in the α3 domain (residues 227 and 229) and the other in the α2 domain (residues 128-136)43,58,59 (Fig. 22.2A). Based on the location of the latter site and molecular dynamic modeling, it has been theorized that the α2 interaction site might function as a folding sensor for C-terminal peptide anchoring in the MHC-I F pocket.60,61 Consistent with this model, polymorphisms within the F pocket can have a profound effect of Tpn dependencies of different MHC-I alleles.62 Based on mutagenesis and structural analyses, a credible model was constructed by Dong et al. of the MHC-I/PLC interactions52 (see Fig. 22.2).
Although Tpn within the PLC is thought to enforce most of the quality control of antigen presentation by MHC-I, there are also adjunct pathways. For example, the sensor UGT1 that adds a terminal glucose to unfolded ER proteins for rebinding CRT has been recently implicated in the quality control of antigen presentation by MHC-I. In support of this conclusion, using in vitro assays with recombinant components, UTG1 was reported to preferentially reglucosylate MHC-I proteins loaded with suboptimal peptides.63 Also, as a post-ER quality control pathway, CRT was reported to use a KDEL-dependent mechanism to recycle suboptimally loaded MHC-I proteins from the early Golgi back the ER for improved peptide binding.64
Viral Immune Evasion of Antigen Presentation
As noted previously, several molecular components of antigen presentation were coopted from physiologic pathways of protein degradation and quality control. Using elegant mechanisms, viruses express diverse proteins that coopt these same pathways of protein degradation and quality control to evade immune detection by CD8 T cells.65 Indeed, the specificity and potency of immune evasion proteins impairing antigen presentation makes them efficacious probes for physiologic pathways of relevance for MHC expression. Most of the well-characterized immune evasion proteins are expressed by deoxyribonucleic acid viruses with large genomes, particular viruses capable of latency or host coexistence. Strikingly, immune evasion proteins block several different steps of the antigen presentation pathway by MHC-I proteins (Fig. 22.3). For example, Epstein-Barr virus and Kaposi sarcoma-associated herpesvirus express proteins during latency that escape cytotoxic T lymphocyte detection by containing sequences that inhibit proteasome processing.66,67,68,69,70 Alternatively, blocking peptide transport by inhibiting TAP function is a commonly used immune evasion strategy. For example, ICP47 of herpes simplex virus binds the cytosolic side of TAP and blocks peptide and not ATP binding,71,72,73,74,75,76 whereas US6 of human cytomegalovirus (HCMV) binds the luminal side of TAP and induces a conformational change resulting in inhibition of ATP hydrolysis
and peptide translocation.77,78,79,80,81 As a third mechanism of TAP blocking, UL49.5 of bovine herpesvirus induces TAP degradation.82 There are also published examples of immune evasion proteins inhibiting Tpn function. For example, US3 of HCMV binds to Tpn and impairs optimization of peptide loading,83,84 whereas E3-19K of AdV inhibits the ability of Tpn to bridge MHC-I with TAP.85
and peptide translocation.77,78,79,80,81 As a third mechanism of TAP blocking, UL49.5 of bovine herpesvirus induces TAP degradation.82 There are also published examples of immune evasion proteins inhibiting Tpn function. For example, US3 of HCMV binds to Tpn and impairs optimization of peptide loading,83,84 whereas E3-19K of AdV inhibits the ability of Tpn to bridge MHC-I with TAP.85
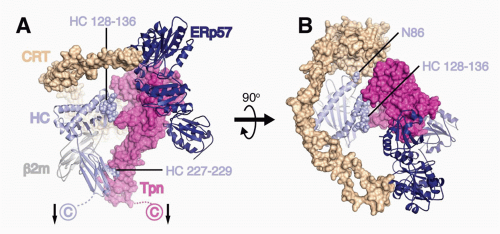 FIG. 22.2. A Model of the Peptide Loading Complex Based on Known Structures and Mutagenesis Data of Interaction Sites.52 Heavy chains (HCs), β2m, and ERp57 are shown as ribbons, while calreticulin (CRT) and tapasin (Tpn) are shown as Connolly surfaces (1.4 Å probe). HCs and Tpn are membrane anchored as indicated with arrows, whereas transmembrane interactions between Tpn and TAP (not shown) attach the phospholipase C (PLC) components to TAP. HC sites predicted by mutagenesis studies to interact with Tpn (residues 227 to 229 and 128 to 136) and CRT (N86) are indicated as spheres. The long flexible P domain of CRT extends above the major histocompatibility complex (MHC)-I peptide binding platform before it noncovalently binds to ERp57, which is covalently attached to Tpn. A: PLC components are shown from the side to highlight Tpn contacts to the HC α2 and α3 domains. B: PLC components shown from above reveal coordinated chaperone binding of both MHCI α-helices near the antigenic peptide’s C-terminus. The figure was rendered by Drs. William McCoy IV and Daved Fremont using Protein Data Bank (PDB) files kindly provided by Drs. Karin Reinisch and Peter Cresswell. The structures of the Tpn/ERp57 complex was published in Dong et al.,52 the structure of CRT was modeled using PHYRE2235 on its homolog calnexin,236 and human leukocyte antigen-A2237 is shown as a representative HC. It should be noted that the HC conformation shown is that attained after peptide occupancy, as the peptide-accessible HC conformation that binds the PLC has not been resolved. |
Immune evasion proteins also employ strategies to misdirect trafficking of MHC-I proteins. For example, the aforementioned E3-19K protein of AdV binds MHC-I proteins and blocks their transport out of the ER.86,87,88,89,90 Interestingly, cowpox virus expresses two different immune evasion proteins targeting MHC-I proteins that function in tandem. Cowpox virus 12 is a TAP function blocker that curtails peptide supply, whereas cowpox virus 203 returns fully assembled MHC-I proteins from the Golgi back to the ER.91,92,93,94 To also misdirect assembled MHC-I proteins, the Nef protein of human immunodeficiency virus and gp48 protein of murine cytomegalovirus shuttle assembled MHC-I proteins from the Golgi to the lysosome.95,96,97
Interestingly, the aforementioned immune evasion proteins for the most part do not have cellular homologs, making it difficult to track their evolution. Striking exceptions to this generality are the viral ER ubiquitin ligases called the viral MARCH (membrane-associated RING-CH) proteins. The extensive family of MARCH proteins includes viral and cellular homologs that share a transmembrane orientation and a highly conserved atypical RING domain that confers E3 ubiquitin ligase activity.98,99,100 Viral proteins mK3 of MHV68, and proteins kK3 and kK5 of Kaposi sarcoma-associated herpesvirus that function as immune evasion proteins were the founding members of the MARCH protein family.101,102,103 Mechanistically, mK3 binds to TAP and awaits the entry of MHC-I into the PLC after which mK3 ubiquitinates the cytosolic tail of MHC-I HCs and thus induces their dislocation to the cytosol and degradation in the cytosol (ie, ERAD).104,105,106,107 Of note, the extensively studied US2 and US11 proteins of HCMV also target ERAD of MHC-I proteins by recruiting cellular E3 ligases.108,109,110,111,112,113 Indeed, studies of US2, US11 and mK3 continue to provide molecular insights into various mechanisms by which ERAD substrates are detected in the ER and dislocated to the cytosol.114,115 In contrast to ERAD, the kK3 and kK5 MARCH ligases of Kaposi sarcoma-associated herpesvirus induce endocytosis and lysosomal degradation of MHC-I proteins. Interesting, kK3 like mK3 appears to ubiquitinate only MHC-I proteins, whereas kK5 targets other surface receptors including the T cell costimulation molecule CD86 (B7.2) as well as natural killer cell ligands, MHC class I-related chain A or B (MICA, MICB), and newly defined ligand for NKp80, activationinduced C-type lectin (AICL).116 Inhibition of natural killer responses is of importance to the virus because downregulation of MHC-I proteins renders cells susceptible to natural killer cell lysis. Mechanistic studies of viral MARCH
proteins have provided several molecular insights into ubiquitin-dependent degradation pathways of particular relevance to antigen processing. In addition, there are about 10 cellular MARCH protein homologs in humans and mice, many of which detect immune receptors.98,99,100 For example, as mentioned in the MHC-II section, MARCHI regulates MHC-II expression in B cells and dendritic cell (DC) subsets.117,118,119 Thus, viral MARCH proteins were stolen from the host and adapted by the virus to function as immune evasion proteins using similar mechanisms.
proteins have provided several molecular insights into ubiquitin-dependent degradation pathways of particular relevance to antigen processing. In addition, there are about 10 cellular MARCH protein homologs in humans and mice, many of which detect immune receptors.98,99,100 For example, as mentioned in the MHC-II section, MARCHI regulates MHC-II expression in B cells and dendritic cell (DC) subsets.117,118,119 Thus, viral MARCH proteins were stolen from the host and adapted by the virus to function as immune evasion proteins using similar mechanisms.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


