Fig. 7.1
Representative structures of systemic antifungal agents
Limited absorption of the drug at elevated gastric pH.
Lack of an intravenous (IV) formulation.
Requirement for extensive cytochrome P450 biotransformation before elimination, resulting in a high propensity for drug–drug interactions.
Dose-related gastrointestinal, hepatic, and adrenal toxicity.
Limited penetration into anatomically privileged sites such as cerebrospinal fluid (CSF).
In an attempt to address these limitations, a new chemical group of azoles (triazoles) were developed with improved physiochemical characteristics and spectrum of activity. Fluconazole, introduced in the early 1990s, could be administered intravenously or orally, had predictable pharmacokinetics, excellent oral bioavailability, and improved penetration into anatomically restricted sites such as the vitreous humor and CSF. Importantly, fluconazole was well tolerated and was associated with few serious drug interactions in critically ill patients. As a result, fluconazole quickly became one of the most widely prescribed antifungal for superficial and life-threatening infections due to yeasts [1]. The lack of activity against opportunistic molds (e.g., Aspergillus, Fusarium, and the Mucorales) and intrinsic resistance among some non-albicans Candida species (i.e., Candida glabrata and Candida krusei), however, created a need among severely immunocompromised patients for broader-spectrum alternatives. The development of itraconazole and the broader-spectrum triazole derivatives, voriconazole and posaconazole and more recently isavuconazole, has largely addressed the spectrum limitations of fluconazole among these high-risk patients. Yet, these broader-spectrum triazoles still carry a potential for cross-resistance with fluconazole [2] and exhibit more complex pharmacokinetic profiles, and a higher propensity for drug interactions. Hence, less toxic alternatives to triazole antifungal therapy would be desirable in critically ill patients, especially patients at higher risk of pharmacokinetic drug–drug interactions.
The final milestone of antifungal drug discovery during the twentieth century was the identification and development of the echinocandins; lipopeptide molecules that inhibit glucan synthesis leading to damage of the fungal cell wall [3]. Due to the importance of the cell wall in fungal survival and the lack of this target in mammalian cells, echinocandins were predicted to be well-tolerated antifungal agents with little collateral toxicity in humans. Yet, the first echinocandin tested in humans, cilofungin, had to be abandoned prior to large-scale clinical trials due to difficulties in its preparation and the toxicity of its IV formulation [4]. Subsequent semisynthetic echinocandin derivatives demonstrated improved solubility and potency and were well tolerated even at high dosages. In 2001, caspofungin became the first echinocandin approved by the Food and Drug Administration (FDA) for the treatment of invasive fungal infections in humans. Two other echinocandin derivatives with a similar spectrum as caspofungin, anidulafungin and micafungin, have subsequently been approved for human use.
Although the arrival of new antifungal agents has clearly advanced the management of invasive fungal infections, treatment failures still occur, and some patients do not tolerate particular antifungal agents due to hypersensitivity reactions, renal or hepatic toxicity, or the potential for serious drug interactions. Therefore, no single antifungal agent is appropriate for all patients for any given mycoses. Moreover, breakthrough infections with pathogens that have acquired intrinsic resistance to multiple antifungal classes have become more common with prolonged treatment courses and improved survival of chronically immunocompromised hosts. This trend has created an urgent need for laboratory support in the treatment of invasive fungal infections including: (a) the rapid identification of fungal pathogens to the species level, and (b) in vitro susceptibility testing of clinical isolates to guide the selection of antifungal therapy. This chapter reviews key components of antifungal pharmacology with a special emphasis on systemic antifungal agents and common patterns of resistance among opportunistic mycoses in humans.
Targets of Antifungal Therapy
Despite differences in the sterol composition of the cell membrane and the presence of a cell wall, much of the cellular machinery of fungi shares remarkable homology to mammalian cells. Consequently, development of drugs that selectively target pathogenic fungi without producing collateral damage to mammalian cells is a daunting pharmacological challenge. Not surprisingly, many of toxicities and drug interactions observed with contemporary antifungal therapies can be attributed to “nonselective” interactions with homologous enzyme or cell membrane systems found in mammalian host cells [5].
With the exception of flucytosine, currently available systemic antifungals act primarily through direct or indirect interactions with the fungal cell wall or plasma membrane, and the fungal membrane sterol, ergosterol, and its biosynthetic pathways (Fig. 7.2). The fungal cell envelope has several properties that make it an ideal target for antifungal therapy [4]:
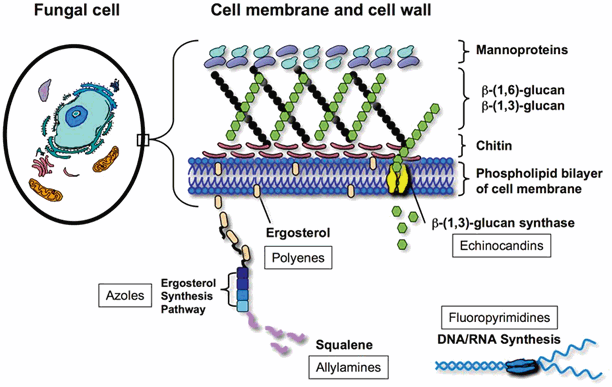
Fig. 7.2
Targets of antifungal therapy
In contrast to the cholesterol-rich cell membranes of mammalian cells, the predominant cell membrane sterol in pathogenic fungi is ergosterol. Indirect or direct targeting of ergosterol results in selective toxicity to the fungal cells.
Mammalian cells lack a true cell wall. Drugs that target synthesis of the fungal cell wall have a low potential to cause collateral toxicity in mammalian cells.
The cellular wall and membranes are important for ion exchange, filtration, and are a critical area for localization of enzymes involved in the metabolism and catabolism of complex nutrients [6]. Drugs that disrupt growth of the cell membrane and wall produce a number of pleiotropic effects that can arrest fungal growth.
Polyene Mechanisms of Action
Polyene antifungals (amphotericin B) bind to ergosterol, the principal sterol in the fungal cell membrane, disrupting the structure of the fungal cell membrane to the point of causing leakage of intracellular contents. Although this binding typically results in rapid cell death, the precise mechanism of fungicidal activity remains unknown. Structurally, the fungal sterol, ergosterol, exhibits a more cylindrical three-dimensional structure than the mammalian sterol, cholesterol, which largely explains the greater affinity of amphotericin B binding to ergosterol (Fig. 7.3; [4]). However, amphotericin B undoubtedly binds to cholesterol in mammalian cell membranes; a mechanism that could account for the direct toxicity of the drug to the distal tubules of the kidneys [5].
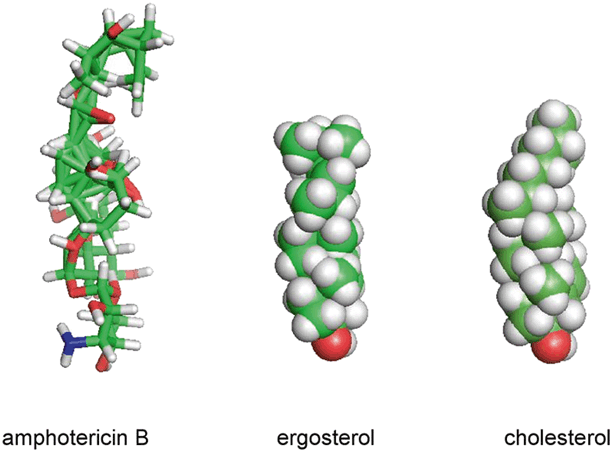
Fig. 7.3
Amphotericin B, ergosterol, and cholesterol visualized in three-dimensions
Azole Mechanisms of Action
In contrast to the direct interactions of the polyene antifungals with ergosterol, azole antifungals indirectly affect the fungal cell membrane through inhibition of ergosterol biosynthesis. Azole antifungal compounds inhibit cytochrome P-450 sterol 14α-demethylase (Erg11p or CYP51p depending on nomenclature), an enzyme that catalyzes the oxidative removal of 14α-methyl group of lanosterol in the ergosterol biosynthetic pathway. Inhibition of 14α-demethylase by azoles results in an accumulation of 14α-methylated sterols in the cytoplasmic membrane, which disrupt phospholipid organization, impair membrane-bound enzyme systems such as ATPase and enzymes of the electron transport system; thus arresting fungal cell growth. CYP51p enzyme binding is accomplished through coordination of the triazole N3 or imidazole N4 of the azole ring with the cytochrome P-450 heme target site, while the remainder of the drug molecule binds to the apoprotein in a manner dependent on the individual structure of the azole (Fig. 7.4; [4]). Differences in the exact conformation of the active site between fungal species and drug structure largely define the spectrum of each agent. For molecules derived from the ketoconazole pharmacophore (e.g., itraconazole, posaconazole), extension of side chain enhances binding of the azole to the P-450 apoprotein, and expands the potency and spectrum against both yeast and filamentous fungi. For molecules derived from fluconazole (e.g., voriconazole) inclusion of an α-O-methyl group confers activity against Aspergillus and other filamentous fungi [7].
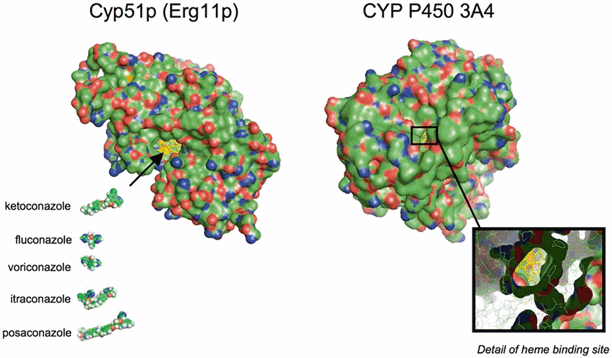
Fig. 7.4
Cyp51p and CYP-P450 3A4 visualized in three dimensions highlighting the hemebinding site for the imidazole (ketoconazole) and triazole rings
One drawback of targeting fungal CYP-450 enzymes involved in ergosterol biosynthesis is the homology the fungal enzyme systems share with mammalian CYP-450 enzymes involved in drug metabolism (Fig. 7.4). Indeed, all azoles inhibit to varying degrees the mammalian CYP-P450 enzymes in humans involved in drug metabolism [8]. Azole therapy can predispose patients to a number (i.e., > 2000 theoretical) of pharmacokinetic drug–drug interactions when these antifungals are administered concurrently with drugs that are either substrates or inducers of CYP-P450 enzymes in humans [9]. Unfortunately, modifications of the azole pharmacophore designed to enhance binding to fungal CYP51p frequently enhances binding of mammalian CYP-P450 enzymes. Therefore, improvement in the spectrum of azole antifungals is often accompanied by an increased potential for drug interactions.
Allylamine Mechanisms of Action
Similar to azoles, allylamines inhibit ergosterol biosynthesis prior to 14 α-demethylase by inhibition of the squalene monooxygenase (formally epoxidase). This enzyme is responsible for conversion of squaline to squalene epoxide, a precursor of lanosterol in the ergosterol biosynthetic pathway. Following exposure to allylamines such as terbinafine, the fungal cell membrane accumulates squalene while becoming deficient in ergosterol resulting in arrest of cell growth. Although allylamines do not appear to produce the same degree of cross-inhibition of mammalian CYP-P450 enzymes as azole antifungals, strong inducers of mammalian CYP-P450 enzymes such as rifampin still increase the metabolism of squalene monooxygenase inhibitors such as terbinafine.
Echinocandin Mechanisms of Action
Of the currently available antifungal agents, only one class of agents, the echinocandins, are known to specifically target fungal cell wall synthesis. Echinocandins inhibit the synthesis of 1,3-β-d-glucan polymers, which serve as essential cross-linking structural components of the cell wall. Depletion of 1,3-β-d-glucan polymers leads to a structurally impaired cell wall, osmotic instability, and lysis in rapidly growing cells. The presumed target of the echinocandins is thought to be β-1,3-d-glucan synthase, although formal proof of this target in pathogenic fungi has been complicated by technical difficulties in studying the membrane-bound protein complex. In Saccharomyces cerevisiae, where the enzyme complex has been best studied, the echinocandins are known to bind to the Fks1p component of the two proteins (Fks1p and Fks2p) regulated by the guanosine triphosphate (GTP)-binding peptide, Rho1p, that comprise the transmembrane β-1,3-d-glucan synthase complex [10].
The degree of β-1,3-d-glucan polymerization in the cell wall and expression of the β-1,3-d-glucan synthase target chiefly defines the spectrum and lethality of the echinocandins in pathogenic fungi. In Candida species, the fungal cell wall is rich in β-1,3-d-glucans, and the enzyme complex is highly expressed during rapid cell growth. Hence, echinocandins exhibit fungicidal activity against most rapidly growing Candida species. However, echinocandins lack clinically useful activity against Cryptococcus neoformans due to the limited use of β-1,3-d-glucan in the cell wall of this species [11]. Among hyaline molds, the cell wall of Aspergillus species contain the greatest degree of β-1,3- and β-1,6-d-glucan polymers. The β-1,3-d-glucan synthase complex, however, is expressed predominantly on the growing apical tips of the hyphae. Therefore, echinocandins kill only the growing hyphal tips of the fungus, resulting in abnormal, hyperacute branching and aberrant growth, with minimal effects on the viability of subapical components (Fig. 7.5; [12]). Other filamentous fungi, such as Fusarium species and Mucorales, utilize α-1,3-glucans in the cell wall matrix and chitosan polymers [11]. As such, echinocandins lack pronounced activity against these opportunistic fungi.
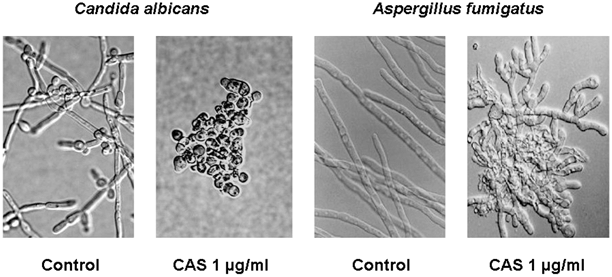
Fig. 7.5
In vitro effects of caspofungin (CAS) on fungal morphology
Pyrimidime Mechanisms of Action
Flucytosine (5-fluorocytosine, 5-FC) works as an antifungal agent through conversion to 5-fluorouracil within fungal cells (Fig. 7.2). Once inside cells, fluorouracil inhibits thymidylate synthase, a key enzyme in DNA synthesis and incorporates into RNA causing premature chain termination. For flucytosine to be effective, it must be internalized and converted to the active fluorouracil form through the activity of two enzymes, cytosine permease and cytosine deaminase. Mammalian cells and many filamentous fungi lack the enzymes for conversion and intracellular concentration of flucytosine, therefore the activity of the drug is most predictable for pathogenic yeast. In humans, however, resident intestinal flora may convert flucytosine to fluorouracil, resulting in nausea, vomiting, diarrhea, and bone marrow suppression [5].
Antifungal Resistance
Antifungal resistance is a broad concept describing the failure of a fungal infection to respond to antifungal therapy. Resistance has been traditionally classified as either primary (intrinsic; i.e., present before exposure to antifungal) or secondary (acquired; i.e., that which develops after antifungal exposure owing to stable or transient genotypic alterations) [13, 14]. A third type of antifungal resistance could be described as “clinical resistance,” which encompasses progression or relapse of infection by a pathogenic fungus that appears, by laboratory testing, to be fully susceptible to the antifungal used to treat the infection. Clinical resistance is most commonly a result of persistent and profound immune defects (e.g., acquired immunodeficiency syndrome (AIDS), neutropenia, graft vs. host disease and its treatment) or infected prosthetic materials (i.e., central venous catheters) which become encased in protective biofilm, thus, limiting drug activity [13, 14]. In some cases, suboptimal drug concentrations at the site of infection, resulting from poor drug absorption, drug interactions, or infrequent dosing, may contribute to clinical resistance.
Primary or secondary antifungal resistance can arise through a number of complex mechanisms and may be expressed over a wide phenotypic spectrum [13]. At one extreme, fungi may be susceptible to the effects of an antifungal agent but growth may not be completely inhibited in vitro. This so-called trailing growth may be observed for antifungals during laboratory testing (particularly azoles and flucytosine) even at high concentrations, but is generally considered an artifact of testing methods and not reflective of clinically relevant resistance. Similarly, some echinocandins may exhibit a paradoxical attenuation of activity at higher drug concentrations without clear evidence of diminished drug activity at higher dosages in animal models or patients. Heterogeneous resistance, the presence of subpopulations of fungal cells with varying degrees of resistance to an antifungal agent in a susceptible population, may indicate an increased propensity for the development of antifungal resistance. This type of resistance may not be detected unless specialized testing methods are used in the laboratory. Inducible or transiently expressed (epigenetic) antifungal resistance mechanisms have also been described in fungi, but little is known about the clinical significance of these resistance patterns in human infections [13]. The other extreme in the phenotypic expression of antifungal resistance is represented by isolates with stable and persistent growth even at high antifungal concentrations. It is important to note that most studies of antifungal resistance focus on isolates with a stable resistance phenotype. Molecular mechanisms of resistance have been best described in Candida albicans isolates recovered from AIDS patients with chronic, recurring fluconazole-refractory oropharyngeal candidiasis [14]. The chronic nature of these mucosal infections allow the longitudinal collection of serial, matched Candida isolates that exhibit progressively stable, higher degrees of resistance to antifungals. By contrast, acute bloodstream candidiasis, aspergillosis, or other less common life-threatening mycoses do not typically allow for the study of serial, matched isolates, thus complicating genotypic–phenotypic correlation of resistance development.
Laboratory Detection of Resistance
Standardization of in vitro tests used to determine the activity of antifungals has been a long process. In 1982, the Clinical Laboratory and Standards Institute (CLSI, formerly National Committee for Clinical Laboratory Standard (NCCLS)) established a subcommittee to assess the need for such testing. It was not until 1985 that the first report of this subcommittee was released. That document, NCCLS M20-CR, Antifungal Susceptibility Testing; Committee Report, was based on responses from hospitals and reference laboratories from across the nation. The committee found that approximately 20 % of the laboratories that responded were, in fact, conducting antifungal susceptibility testing. Many methods existed, but most of the respondents utilized a broth method and were testing yeast only. Comparison testing of isolates between collaborating laboratories was unacceptably low.
Based on this study, the decision was made to develop a standardized method; the goal being not to correlate between isolate and patient outcome but rather between laboratories. Methods that existed included broth, agar, and disk diffusion. The committee decided that the standard method should be a macrobroth dilution method and that only a synthetic medium should be chosen. Several centers collaborated and a preliminary method was introduced in 1992, M27-P [15]. Parameters were set to include medium, inoculum preparation and size, incubation temperature and duration, and endpoint criteria. The procedure has been refined and is now an approved method, M27-A3 [16]. Subsequent publications include M38-A2 [17], M44-A [18], and M51-A [19]. M38-A2 utilizes similar methods for mold testing, while M44-A/M51-A provides guidelines for disk diffusion testing of both yeast and molds. As a result of these approved methods, commercially available kits now allow clinical microbiology laboratories to perform testing in-house, rather than sending isolates off for reference testing. Prior to doing this, however, laboratories should have sufficient requests for this testing to ensure the volume of work needed to maintain accuracy and reproducibility.
Interpretive guidelines, last revised in 2012, have only been established for anidulafungin, caspofungin, micafungin, fluconazole, and voriconazole against specific Candida species. Echinocandin break points are available for C. albicans, C. glabrata, C. tropicalis, C. krusei, C. parapsilosis, and C. guilliermondii, while fluconazole break points are available only for C. albicans, C. glabrata, C. parapsilosis, and C. tropicalis. Voriconazole break points are given for C. albicans, C. krusei, C. parapsilosis, and C. tropicalis. Categories for the echinocandins include susceptible (S), intermediate (I), and resistant (R), while those for the azoles include susceptible (S), susceptible dose dependent (SDD), and resistant (R). The susceptible dose-dependent category relates to yeast testing only and is not interchangeable with the intermediate category used in bacterial and echinocandin break points. This category is in recognition that yeast susceptibility is dependent on achieving maximum blood levels. By maintaining blood levels with higher doses of antifungal, an isolate with an SDD endpoint may be successfully treated with a given azole ([16]; Table 7.1).
Table 7.1
Updated CLSI clinical break points for Candida sp.
Organism | Reportable reading conditions | Break points | |||
|---|---|---|---|---|---|
Susceptible | SDD | Intermediate | Resistant | ||
C. albicans | |||||
Anidulafungin | 24-h 50 % | ≤ 0.25 | – | 0.5 | ≥ 1 |
Caspofungin | 24-h 50 % | ≤ 0.25 | – | 0.5 | ≥ 1 |
Micafungin | 24-h 50 % | ≤ 0.25 | – | 0.5 | ≥ 1 |
Fluconazole | 24-h 50 % | ≤ 2 | 4 | – | ≥ 8 |
Voriconazole | 24-h 50 % | ≤ 0.125 | 0.25–0.5 | – | ≥ 1 |
C. parapsilosis | |||||
Anidulafungin | 24-h 50 % | ≤ 2 | – | 4 | ≥ 8 |
Caspofungin | 24-h 50 % | ≤ 2 | – | 4 | ≥ 8 |
Micafungin | 24-h 50 % | ≤ 2 | – | 4 | ≥ 8 |
Fluconazole | 24-h 50 % | ≤ 2 | 4 | – | ≥ 8 |
Voriconazole | 24-h 50 % | ≤ 0.125 | 0.25–0.5 | – | ≥ 1 |
C. tropicalis | |||||
Anidulafungin | 24-h 50 % | ≤ 0.25 | – | 0.5 | ≥ 1 |
Caspofungin | 24-h 50 % | ≤ 0.25 | – | 0.5 | ≥ 1 |
Micafungin | 24-h 50 % | ≤ 0.25 | – | 0.5 | ≥ 1 |
Fluconazole | 24-h 50 % | ≤ 2 | 4 | – | ≥ 8 |
Voriconazole | 24-h 50 % | ≤ 0.125 | 0.25–0.5 | – | ≥ 1 |
C. glabrata | |||||
Anidulafungin | 24-h 50 % | ≤ 0.125 | – | 0.25 | ≥ 0.5 |
Caspofungin | 24-h 50 % | ≤ 0.125 | – | 0.25 | ≥ 0.5 |
Micafungin | 24-h 50 % | ≤ 0.06 | – | 0.125 | ≥ 0.25 |
Fluconazole | 24-h 50 % | – | ≤ 32 | – | ≥ 64 |
Voriconazole | 24-h 50 % | – | – | – | – |
C. krusei | |||||
Anidulafungin | 24-h 50 % | ≤ 0.25 | – | 0.5 | ≥ 1 |
Caspofungin | 24-h 50 % | ≤ 0.25 | – | 0.5 | ≥ 1 |
Micafungin | 24-h 50 % | ≤ 0.25 | – | 0.5 | ≥ 1 |
Fluconazolea—Resistant | 24-h 50 % | – | – | – | – |
Voriconazole | 24-h 50 % | ≤ 0.5 | 1 | – | ≥ 2 |
C. guilliermondii | |||||
Anidulafungin | 24h-50 % | ≤ 2 | – | 4 | ≥ 8 |
Caspofungin | 24h-50 % | ≤ 2 | – | 4 | ≥ 8 |
Micafungin | 24h-50 % | ≤ 2 | – | 4 | ≥ 8 |
In addition to becoming species specific, break points were also lowered considerably from previous recommendations. All species of Candida were considered susceptible to fluconazole if the MIC was ≤ 8 µg/ml. This break point was decreased to ≤ 2 µg/ml for C. albicans, C. parapsilosis, and C. tropicalis. C. krusei is considered resistant despite the in vitro MIC and C. glabrata is not considered susceptible, but rather SDD at ≤ 32 µg/ml and resistant at ≥ 64 µg/ml. MIC numbers for voriconazole also dropped significantly, from a previous susceptible break point of ≤ 1 µg/ml for all Candida spp., to ≤ 0.125 µg/ml for C. albicans, C. parapsilosis, and C. tropicalis, ≤ 0.5 µg/ml for C. krusei, and a removal of all break points for C. glabrata. The break points for echinocandins also changed for most species. C. parapsilosis and C. guilliermondii remain susceptible at ≤ 2 µg/ml, but C. albicans, C. glabrata, C. tropicalis, and C. krusei susceptibility decreased from ≤ 2 µg/ml to 0.06–0.25 µg/ml depending on the drug–species combination.
One important problem with any approach toward in vitro susceptibility testing is the correlation of the minimum inhibitory concentration (MIC) with patient outcome. Some assumptions can be made, however, about MIC and patient outcome. Rex and Pfaller proposed the “90–60 Rule” as a general guide for establishing clinically relevant interpretative break points for resistance [20]. This rule states that infections caused by isolates that have MICs considered susceptible respond favorably to appropriate therapy approximately 90 % of the time, whereas infections caused by isolates with MICs considered resistant respond favorably in approximately 60 % of cases.
Mechanisms of Resistance
Many aspects of antifungal resistance are still poorly understood, particularly with respect to the regulation and expression of resistance mechanisms following exposure to antifungal agents (secondary resistance). Nevertheless, advances in molecular biology and genome sequencing of pathogenic fungi have yielded progress in our understanding of common mechanisms leading to antifungal resistance. These mechanisms can be grouped into five general categories (Fig. 7.6):
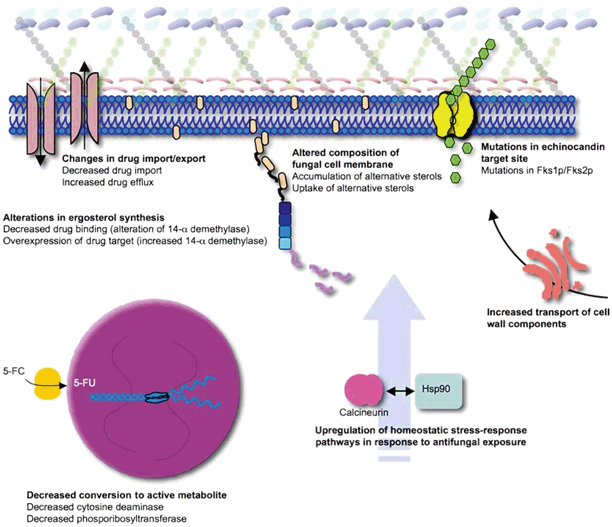
Fig. 7.6
Mechanisms of antifungal resistance
Decreased drug import or increased drug exportation (efflux pumps)
Alteration in drug target binding site
Changes in biosynthetic pathways (particularly sterol synthesis) that circumvent or attenuate the effects of antifungal inhibition
Alterations in intracellular drug processing
Upregulation of homeostatic stress-response pathways to deal with antifungal-associated damage
It is important to note that multiple resistance mechanisms are often expressed simultaneously following antifungal exposure and that a single mechanism is unlikely to result in a resistant strain. Depending on the mechanisms concurrently expressed, cross-resistance may, or may not, be observed between different antifungals. Whole-genome expression profiles of C. albicans have revealed transient upregulation of several resistance mechanisms (e.g., ergosterol biosynthesis—ERG 3, ERG11; efflux pumps—CDR1, CDR2) following a single exposure to azole antifungals [21]. Development of resistance in longitudinally collected clinical strains in patients who fail progressively higher dosages of antifungals generally demonstrate a gradual accumulation of several alternations that result in a detectable resistance [13].
Changes in drug importation and exportation are probably the most common mechanisms associated with primary and secondary antifungal resistance [13]. Decreased drug importation is consistently associated with primary resistance to flucytosine and azoles antifungals. For example, poor uptake of flucytosine due to alterations in cytosine permease or decreased availability of this enzyme, largely account for the limited spectrum of this agent against opportunistic molds. Similarly, differences in azole susceptibility between fluconazole and itraconazole against C. krusei have been reported to be more closely associated with intracellular accumulation than differences in drug-binding affinity to the 14 α-demethylase target [22]. Drug importation may also be affected by the sterol composition of the plasma membrane. Several studies have demonstrated that when the ergosterol component of the membrane is altered in favor of other 14 α-methyl sterols, there is a concomitant permeability change in the membrane to drugs, and a decrease in membrane fluidity [13].
Similar to other eukaryotic cells, fungi are known to contain two types of efflux pumps that contribute to drug resistance: ATP-binding cassette (ABC) transporters and major facilitators (MF). Overexpression of the ATP-dependent ABC transporters typically confers a multidrug resistance phenotype. In contrast, MF pumps, which expel antifungal though proton motive force (H+ gradient across membrane), have a much narrower spectrum of substrate specificity. In C. albicans, overexpression of ATP-dependent efflux pumps CDR1 and CDR2 confer cross resistance to all azole antifungals [13]. In contrast, overexpression of MF pump MDR1 effects only the accumulation of fluconazole and does not result in cross-resistance to itraconazole or ketoconazole. Overexpression of ATP-dependent efflux pumps is the most prevalent mechanism of efflux-mediated resistance reported in clinical isolates [13]. Recently, overexpression of ABC transporters was reported to confer a degree of cross-resistance between azoles and echinocandins in a laboratory strain of C. albicans [23].
Besides drug efflux, the most common mechanism associated with antifungal resistance involves changes in the binding site of the drug. Several genetic alternations in ERG11, the gene encoding 14 α-demethylase, have been attributed to decreases in azole activity, including point mutations that result in changes in the active pocket site or overexpression of ERG11. Similar alterations in other enzymes of the ergosterol biosynthetic pathway, particularly ERG 3 (C-5-sterol desaturase), which is upregulated with inhibition of 14 α-demethylase, have also been documented in azole-resistant clinical strains. Binding site alteration is also likely to be an important mechanism of echinocandin resistance. Point mutations in the FKS1 gene have been linked with clinical failure of echinocandins in the treatment of bloodstream infections and deep-seated candidiasis, especially among C. glabrata [3, 24, 25].
Changes in the target expression in the ergosterol biosynthetic pathway alter the fungal cell membrane sterol content. Substitution of alternative sterols for ergosterol, or alterations in the sterol:phospholid ratio in the cell membrane can decrease intracellular accumulation of azoles and reduce the binding of amphotericin B to the cell membrane. Indeed, most polyene-resistant yeasts recovered from patients with clear microbiological failure on amphotericin B exhibit decreased ergosterol concentrations in their fungal cell membranes. Several studies have even suggested that pathogenic fungi can scavenge free sterols for the cell membrane, including cholesterol, resulting in resistance to polyene and azole antifungals [26].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


