Fig. 1
VWF functions: After the platelets detect any defect in vascular wall, shear rate leads to the initial arrest and attachment of platelet at the site of injury. Shear-induced conformational changes in vWF may contribute to the regulation of vWF binding to platelet GPIbα. Excess of the large vWF multimers in circulation results in platelet clumping afterwards
Moreover, vWF has been proposed as a novel marker for the increased risk of re-infarction and mortality in survivors of myocardial infarction [12–14]. vWF has also been associated with thromboembolism, metastasis and angiogenesis [15]. In addition, a marked increase in vWF levels was found in HIV patients with a history of thrombosis [17]. The above studies, however, have made vWF a remarkable target for research on CVD, atherosclerosis and the rest of thrombotic events.
2 VWF Gene
VWF is encoded on the short arm of chromosome 12 at position 13.3 and comprises 180 kb length and 52 exons [18]. The VWF gene belongs to a family of genes named endogenous ligands. Twin studies have revealed that 66 % of the discrepancy of vWF level underlies a genetic influence and 30 % of the genetic variance is because of the effect of ABO blood group. Naturally occurring genetic differences among individuals in the VWF gene are linked with vWF levels. These includes polymorphic variations in the 5′-regulatory region of hemostatic factors, which can contribute to the level of vWF presence in plasma and eventually the risk of thrombotic events. In addition, the presence of three single nucleotide polymorphisms (SNPs) within the regulatory region of vWF gene at nucleotides −1234C/T, −1185A/G and −1051G/A demonstrated a significant association with plasma levels of vWF:Ag in a population of normal group O blood donors [19]. The 1793C/G polymorphism is in the promoter region of the gene and the G allele is linked with greater levels of vWF antigen, which is more noticeable after the age of 40. In addition, endothelial cell-derived nuclear proteins preferred to bind to certain alleles at these sites [20]. The association of 1793C/G polymorphism with the incidence of ischemic stroke has also been reported in a number of studies [21, 22].
3 VWF Biosynthesis
The location of vWF biosynthesis and secretion is consistent with its biological function, which is in a close association with that of platelet. It is primarily synthesized and stored in endothelial cells, megakaryocytes and platelet precursors in the bone marrow. vWF as a large multimeric glycoprotein is present in blood plasma, platelet α-granules, sub-endothelial connective tissue and endothelium with a plasma concentration of approximately 5–10 μg/ml [23]. The plasma form of smaller size is corresponding to the dimer, with an approximate size of 500,000 Da.
3.1 Synthesis of vWF in Endothelial Cells
Normally, vWF is synthesized as pre-pro-vWF in endothelial cells. This immature form of protein encompasses the signal peptide, pro-peptide and mature subunit. The protein goes through a process during which the signal peptide is removed, and dimerization and multimerization occur, respectively [24]. The pro-peptide, a tag for the protein, is stored in the storage granules and can be detached before releasing into blood, where it is known as von Willebrand antigen II (vWAgII). In the endothelial cells, vWF is stored in specialized rode-like granules identified as Weibel–Palade bodies (WPB), which are assumed to be a derivative of Golgi apparatus. It is worth noting that vWF multimerization itself is confirmed to trigger WPB formation. VWF, therefore, is responsible for the cigarette shape of WPBs [25]. These constructions are able to rapidly respond to alteration in the integrity of the cells covering the vessel walls. VWF, however, is synthesized in the vascular endothelial cell through both constitutive and stimulated release (Fig. 2). This major part of vWF (85 % of all vWF) may either constitutively be secreted in plasma or deposited in the WPBs. Moreover, circulating plasma vWF mostly originates from endothelial cells, while platelet vWF might be released through platelet activation. The role of plasma vWF in the hemostatic process is more dominant than platelet vWF (Figs. 2 and 3). The modification is a vital occasion that may affect different steps from biosynthesis to function and finally to degradation of a protein. VWF glycosylation as a posttranslational modification results in the covalent attachment of carbohydrate structures to the protein backbone.
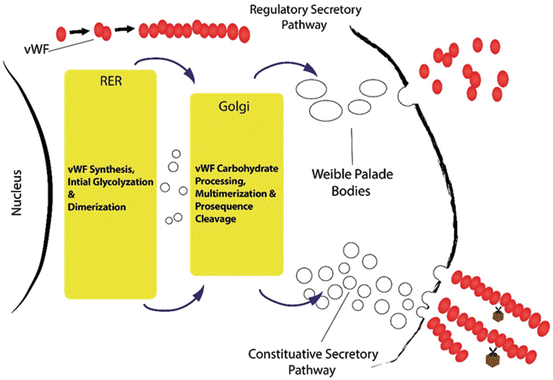
Fig. 2
VWF from biosynthesis to release: Glycosylation is a posttranslational modification that results in the attachment of carbohydrate to the protein backbone. VWF may be either constitutively secreted in plasma or regulated & deposited in the Weibel-Palade bodies
Thus vWF goes through several modifications, including N-linked and O-linked glycosylation (Fig. 3). O-glycosylation proceeds in the Golgi following N-linked glycosylation, protein folding, and oligomerization. The process is initiated by transferring of N-acetylgalactosamine (GalNAc) from UDP-αGalNAc to a serine or threonine, followed by addition of monosaccharides via specific transferases [26]. In addition, the cellular origin of protein and the variability of enzymes mediating glycosylation in different cell types are the important elements of glycosylation.
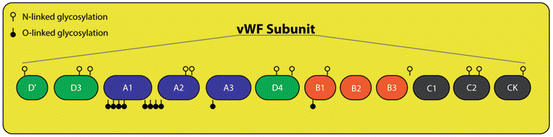
Fig. 3
VWF glycosylation: The location of N-linked and O-linked glycosylation on a vWF subunit
The differences, however, between the vWF secreted constitutively by human endothelial cells and that released from Weibel-Palade bodies after stimulation was explained by Sporn et al. The majority of constitutively secreted molecules were dimeric and contained both pro-vWF and mature subunits as they described. In contrast the vWF released by the calcium ionophore A23187 or thrombin consisted of mature subunits of very large multimers. These large subunits have been recognized to play a very important task. They are more active in platelet binding assays in vitro, and their absence in vivo results in a bleeding disorder. Weibel-Palade bodies though contain a special concentrated subclass of very large and biologically potent vWF multimers [27].
3.2 Synthesis of vWF in Platelet
VWF is also synthesized by the megakaryocyte, which includes 10–25 % of the total amount of vWF antigen present in plasma. However, analysis of reduced vWF purified from human megakaryocytes has demonstrated a subunit composition comparable to that of endothelial cell-derived vWF. Studies have also shown that platelet vWF is placed within the α-granules, as detected by immune-fluorescence study [28], which can be released upon platelet activation. Nevertheless the storage of α-granules may be provided via both biosynthesis at the megakaryocyte and uptake of plasma proteins. This may occur at the megakaryocyte and/or platelet stage [29]. Release of platelet vWF is prompted by a variety of agonists, including ADP, collagen and thrombin [30].
4 VWF Structure
VWF, a long multimeric protein, contains several distinct domains, each with specialized functions. This long polypeptide chain has a molecular mass of ~270 kDa and encompasses the identical repeated domains, whose rearrangement looks like a mosaic protein. VWF monomers are dimerized via disulfide bonds [31]. These bonds subsequently turn these 500 kD dimmers into multimers of different sizes, may be more than 10, 000 kD. VWF monomer, is composed of repeated domains (as seen in Fig. 4) of cDNA, which are responsible for the various binding functions of the molecule [32]. The main functional domains are A1 through which vWF binds to the platelet receptor GPIbα and heparin, A3 for attaching to collagen, C1 that has RGD sequence and is recognized by integrins (αIIbβ3 and αvβ3), and D′–D3, which contains the binding site for FVIII [33].
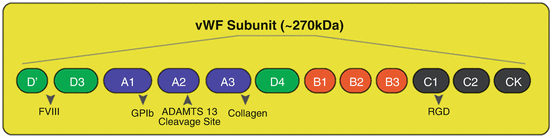
Fig. 4
The main functional domains of vWF subunit: As it is shown, A1 domain binds to the platelet receptor GPIbα & heparin, A2 domain is the cleavage site for ADAMTS13, A3 domain attaches to collagen, C1 domain has RGD sequence and is recognized by integrins (αIIbβ3 and αvβ3), and D′–D3 contains the binding site for FVIII
5 VWF Function
It has been established that active vWF is present only in partial amounts within normal plasma, whereas pathological conditions are usually associated with increased levels of active vWF. Immediately after the platelets detect any defect in vascular wall, a rapid diversion in the blood flow and shear rate leads to the initial arrest and attachment of platelet at the site of injury. This is known as primary homeostasis after which, permanent platelet adhesion and aggregation are formed in the line of vessel endothelium. VWF and platelet vessel-wall interaction promotes GPIbα binding to the halted vWF multimers. This binding promotes the recruitment of circulating platelets to the site of injury, where subendothelium is exposed [34]. Shear rate rising causes the immediate interaction of platelet GPIbα with A1 domain of vWF; an essential adhesive interaction that can tether platelets on the endothelial surface. However, this initial adhesion is an unstable phase in which, vWF plays a key role. The temporary binding may promote a stronger and prolonged adhesion, which is mainly mediated by vWF [35].
Using vWF-A1 knock-in mice, mutations that increase (I1309V) or interrupt (R1326H) platelet receptor glycoprotein Ibα binding were investigated. R1326H mutation into the major site, however, shortened the bond lifetime and resulted in hemostatic and thrombotic defects. In contrast, the I1309V mutation near the minor site prolonged the bond lifetime and developed a type 2B-like vWD phenotype. Nevertheless, combination of these two mutations normalized both bond kinetics and thrombotic properties of vWF. Increased levels of active vWF have been associated with thrombotic complications in several inflammatory conditions, suggesting the role of active vWF in this regard. Additionally, miR-24 has been found to control vWF levels in diabetic patients [36]. These findings highlight the importance of combined biophysical and genetic approaches in identifying potential therapeutic protocols for treatment of bleeding and thrombotic disorders [37].
5.1 VWF and Neutrophil Extracellular Traps (NETs)
NETosis, an innate immune response mediated by neutrophils, has recently been recognized to immobilize microorganisms inside the vasculature in infectious and non-infectious diseases. In fact, NETs are extracellular DNA fibers comprising histones and neutrophil antimicrobial proteins, which might be detected by some markers. Since the markers of extracellular DNA traps were abundantly found in deep vein thrombosis (DVT) [38, 39], NETs were introduced as an exceptional link between inflammation and thrombosis, providing a stimulus and scaffold for thrombus formation. Further investigations have shown co-localization of the DNA network and vWF, indicating the involvement of NETosis mechanism in thrombus formation [40, 41]. A recent study revealed that vWF can bind and immobilize extracellular DNA released from leukocytes. Therefore, vWF might act as a linker for leukocyte adhesion to endothelial cells, but DNA–vWF binding does not affect vWF degradation by ADAMTS13. In contrast, DNA–vWF complexes diminish platelet binding to vWF [42]. These findings, however, have revealed a new feature of vWF biology, introducing vWF and NETs as prospective targets for the development of novel therapeutics for the treatment of venous thrombosis (Fig. 5).
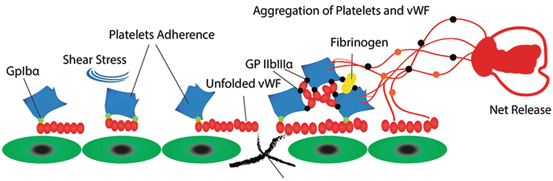
Fig. 5
Involvement of NETs in thrombus formation: in addition to fibrin and von Willebrand factor, NETs characterize a third thrombus scaffold. VWF can bind and immobilize extracellular DNA released from leukocytes at the site of injury
6 Factors Influencing vWF Levels and Activity
The clinical signs of thrombosis perhaps result from the coincidence of multiple adverse genetic and environmental elements. However, a number of parameters may affect vWF levels and activity, such as blood group, genetic variability, acute-phase response, and proteolysis by a disintegrin and metalloprotease with thrombospondin motifs (ADAMTS13, Fig. 6).
6.1 VWF and Blood Group
Plasma vWF levels were determined in more than a thousand blood donors. The results showed that ABO blood group had a significant influence on vWF values, as investigated by quantitative immunoelectrophoresis. The findings also showed that the individuals with blood group O had the lowest mean vWF level than those of non O [43]. VWF antigen levels also showed significant differences between O heterozygotes and non-OO homozygotes [22, 44]. VWF and blood group, however, were related to thrombosis risk, which could be mediated through factor VIII [45].
6.1.1 Updated and Theoretical Rationale for vWF and ABO Interaction
It was though that ABO blood group might modify the synthesis and secretion rate of vWF within endothelial cells. In addition, ABO group may affect vWF plasma clearance rates, which might vary by different types of ABO group [46]. VWF multimers are built up from vWF monomers that contain 12 N-linked and 10 O-linked glycosylation sites. Even though the functions of these glycosylation sites are not fully elucidated, there is some evidence showing that they may protect vWF from proteolytic degradation. This glycosylation may also be essential for dimerization, polymerization and secretion, respectively. The reduced number of terminal sugars on N-linked glycan increases susceptibility of globular vWF to ADAMTS13 proteolysis, thereby reducing plasma vWF levels. ABO antigens have been shown to present on vWF-derived N-linked carbohydrates [41]. Nevertheless, the existence of blood group antigens on vWF O-linked carbohydrates has not been reported. VWF level has, therefore, been demonstrated to be slightly higher for homozygotes (AA or BB) than heterozygotes (AO or BO) [46, 47]. Expectedly, Bombay phenotype showed a lower plasma-vWF levels and an increased proneness to ADAMTS13 proteolysis [48]. In addition, individuals with homozygous secretor genotype (SeSe) showed a higher level of plasma vWF in comparison with non-secretors [49]. Another study, however, demonstrated that the risk of venous thrombosis was not associated with the secretor status [50]. As a result, ABO blood group influences the expression of the vWF involved in hemostasis and accordingly can be linked to various thrombotic events [22, 51–53].
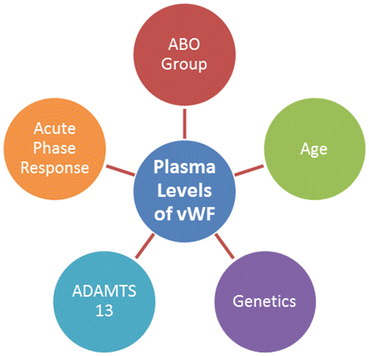
Fig. 6
Factors influence vWF antigen levels and activity: the most important elements affect vWF antigen levels and activity, containing blood group, genetic variability, proteolysis by ADAMTS13 and acute-phase response
6.2 VWF and Aging
The results of multiple regression analysis revealed that age was significantly correlated with vWF levels in each blood group [43]. In addition, G allele, which is linked with higher levels of vWF antigen, is more marked after the age of 40, as previously mentioned. Later studies also confirmed that vWF was increased with age increase [54]. In contrast, plasma levels of ADAMTS13 were reduced by aging in humans. This might indicate an increasing chance of thrombotic events, although the mechanism responsible for this is not accurately clarified [22, 55].
6.3 The Role of a Disintegrin-Like and Metalloprotease with ThromboSpondin Type I Repeats-13 (ADAMTS13)
Since the active large vWF multimers were found to be degraded to smaller forms with less activity, Furlen et al. could reveal a proteolytic activity associated with a high molecular weight protein [56]. The weight of this protein, however, was about 300 kD. Purified vWF was treated with protease. Subsequently, the size, amino acid composition, and amino terminal sequence of the smaller fragments indicated that the cleavage of vWF by the plasma protease was dependent on its conformation and needed calcium ion [57, 58]. Eventually, a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) was defined to mediate the breakdown of vWF. This takes place by the cleavage between tyrosine at position 842 and methionine at position 843 [56, 59]. Decreased vWF-cleaving activity has been reported in a wide range of conditions, including thrombotic thrombocytopenic purpura (TTP), idiopathic thrombocytopenic purpura (ITP), liver cirrhosis, chronic uremia, disseminated intravascular coagulation (DIC), systemic lupus erythematosus (SLE), leukemia, pregnancy, postoperative state, neonatal period, and ageing. Unconjugated bilirubin also inhibited proteolytic cleavage of vWF by ADAMTS13, as it was reported by a recent study [60]. This metalloprotease stretches the vWF A2 domain so that the cleavage site becomes available to hydrolyze the peptide bond within the A2 domain of vWF. Cleavage by ADAMTS13 facilitates vWF release into blood circulation [61]. Next cleavage by ADAMTS13 takes place after vWF release into the blood stream [62], where the elongated conformation occurs. This conformation not only exposes the binding site for platelets in the vWF A1 domain but also expands the vWF A2 domain. Consequently the cleavage site becomes accessible for ADAMTS13. The cleavage site, however, is bordered by the regions of vWF that bind platelet glycoprotein 1b in the A1 domain and collagen [63–65], resulting in two fragments of 176 and 140 kDa. ADAMTS13 cleavage site in vWF is enclosed by two N-linked glycosylation sites (asparagine 752 and asparagine 811) and five O-linked glycosylation sites (threonine 705, 714, 724 and 916, and serine 723). The presence of O, A or B blood groups bordering the site might be the reason for altered values of vWF in various ABO blood groups. It is suggested that this neighboring may influence proteolysis. However, individuals with the lowest levels of ADAMTS13 have shown twice the risk of ischemic stroke compared with individuals with the highest levels of ADAMTS13 [21]. In addition, reduced ADAMTS13 activity significantly exacerbates ischemic brain injury in murine stroke models [66]. Temporary appearance of ultra-large vWF (ULvWF) multimers is evident in the normal plasma of patients after therapeutic agent desmopressin (1-desamino-8-D-arginine vasopressin or DDAVP). ULvWF multimers are also present in the pathological state of diseases such as thrombotic thrombocytopenic purpura (TTP) in which the deficient ADAMTS-13 is a hallmark for the disease [67]. However, the relationship of both vWF and ADAMTS13 with arterial thrombosis has been reviewed. The most studies, however, have shown an association between high vWF levels and arterial thrombosis. Furthermore, a hydrophobic pocket in the Cys-rich domain of ADAMTS13 was recognized, which may interact with hydrophobic pocket in the A2 domain of vWF for its cleavage [68]. Although the infusion of recombinant human ADAMTS13 reduced the infarct size in mice [69], whether ADAMTS13 is a pivotal independent risk factor or not remains unclear.
7 Plasma vWF Versus Platelet vWF
Since von Willebrand factor (vWF) is stored within both the Weibel-Palade bodies and the platelet alpha-granules, it was thought that these two storages should be replaced with each other. The pool of platelet vWF, however, is distinct from plasma-vWF and consists of hemostatically-active high molecular weight multimers. In addition, the glycosylation profile of platelet vWF significantly differs from that of plasma vWF. Furthermore, total sialic acid and galactose expression are little on platelet vWF. ABO blood group carbohydrate determinants do not also exist on the N-linked glycans of platelet vWF. Accordingly, the critical role of vWF glycans is modulating its activity, and functional properties of platelet vWF vary markedly from those of plasma-vWF (Table 1). It has been reported that platelet-vWF could be bound to both GpIIbIIIa and heparin. Nevertheless platelet-vWF binding to platelet GpIbα occurs with a higher affinity in comparison with plasma-vWF [70].
Table 1
The differences between platelet vWF and plasma vWF
Platelet vWF | Plasma vWF |
|---|---|
Higher molecular weight | Lower molecular weight |
Less sialic acid & galactose | More sialic acid & galactose |
Lack of ABO BG carbohydrate | Presence of ABO BG carbohydrate |
Lower affinity to platelet GpIbα | Higher affinity to platelet GpIbα |
Higher affinity to GpIIbIIIα and heparin | Lower affinity to GpIIbIIIα and heparin |
Specific resistance to ADAMTS13 proteolysis | No specific resistance to ADAMTS13 proteolysis |
In addition, both platelet-vWF antigen levels and activity differ significantly between patients with different types of von Willebrand disease (vWD). However, animal model studies have suggested that both types of vWF play important roles in obtaining the primary hemostasis [71]. There was a correlation between the bleeding time and platelet-vWF activity and platelet-vWF Ag. The length of bleeding time though was inversely correlated with the level of platelet vWF activity. The plasma-vWF Ag and activity, however, did not significantly correlate with the bleeding time, indicating that the platelet-vWF plays a greater role in the early stages of hemostasis [72]. Further investigations have demonstrated that plasma vWF and subendothelium support thrombosis, whereas plasma FVIII and platelet vWF are not essential for thrombosis in dogs [73]. Recent findings demonstrated that platelet-vWF with a particularly marked reduction in N-linked sialic acid expression displayed resistance to ADAMTS13 proteolysis. Accordingly, high local concentrations of this type of vWF at the site of injury may further enhance a platelet plug formation that is resistant to ADAMTS13 [74]. The molecular mechanisms responsible for the variances between these two types of vWF are likely to be associated with the differences in the posttranslational modification.
8 VWF and Shear Stress
Platelet adhesion to a thrombogenic surface is highly dependent on the velocity of blood flowing around the site of injury. Since blood consists of concentric layers, the flow rate markedly differs through the lumen. Shear stress is the force per unit area between such layers, which varies through the artery lumen from minimum level at the center-line to maximum level near the wall. In contrast with arterioles, shear rates are generally low in the venous part of the circulation. At a shear rate of >500 s−1, the initial binding of platelets to the vessel wall is mainly mediated by glycoprotein (GP) Ib-V-IX and the von Willebrand factor (vWF). Studies have indicated that vWF principally stimulates platelet activation through an adenosine diphosphate (ADP) release. The investigations have also shown that activation of vWF-GPIbα interacting platelets is triggered by transmembrane Ca2+ [75]. It is reported that under higher shear stress ADP signaling may cause platelet adhesion and activation through P2Y12, a chemoreceptor for ADP, facilitated by immobilized vWF to persistent thrombus formation [76]. The marked shear gradients around stenotic sites such as arteries with advanced atherosclerosis stimulate the endothelial release of vWF and initiate thrombus formation [77]. A recent study identified a role for vWF in transducing hemodynamic forces, indicating that amplified platelet aggregation in stenotic channel is associated with plasma and endothelial vWF. It is thought that vWF goes through a conformational change from a compacted spherical to a prolonged form at high shear stress. It was also suggested that the size of vWF fractions may be varying concomitantly with the shear stress rate [78]. A recent study, moreover, showed that removal of O-linked glycan structures of vWF elevates the flexibility of hinge linker region between the D3 and A1 domains. This assisted vWF unfolding by shear stress and improving its ability to bind collagen and to arrest platelets. The result indicates the important functional role of vWF O-linked glycan structures under shear stress conditions [79]. Furthermore, a recent study showed that in a higher shear stress, the loss of ULvWF multimers was greater and the functional activity of vWF for platelet aggregation was lower [80]. The above results may support the theory of conformational extension for vWF in shear flow (Fig. 7).

Fig. 7
vWF and Shear stress: vWF undertakes a conformational change from a compacted globular to a prolonged form at high shear stress
9 Von Willebrand Factor and Thrombosis
Thrombosis, as the pathological characteristic of hemostatic process, is the foremost cause of death, particularly in the Western society. There are several therapeutic approaches that have been developed to treat this severe clinical complication. Thrombus is structured by numerous elements, including endothelial cells, plasma proteins and shear stress alteration [24]. As vWF plays a critical role in both thrombotic and bleeding events, an elevated plasma level of this factor may predict a thrombotic occurrence but a decreased plasma level may point to a bleeding condition. We investigated the effect of C-reactive protein at moderate levels (the concentrations which are related to atherosclerosis in vivo) on the endothelial cells in vitro. The results exhibited an increased level of vWF, signifying an association between enhancement of vWF levels and risk of thrombosis [12]. Up to now, enoxaparin and Polyethylene glycol (PEG)-hirudin have been used to control vWF rise in cases with unstable angina pectoris [81]. This has been linked with a progressive clinical outcome. Nevertheless, a recent study showed that p-selectin, as an inhibitor of vWF, promoted the resolution of thrombus better than its enoxaparin [82]. Besides, one group identified plasma sodium concentration to increase blood coagulability by affecting the vWF production. They reported that severe hypernatremia reversibly increased both vWF mRNA and vWF secretion in cultured endothelial cells. This was conducted via transcription factor NFAT5, suggesting the involvement of hypertonic signaling in vWF up-regulation. The result however indicated that extracellular sodium enhancement might increase coagulability and risk of thrombosis [83]. Testing the compounds targeting vWF-mediated platelet adhesion points to a favorable bleeding risk profile and higher efficacy compared with traditional antithrombotic drugs. Antibody to vWF, however, inhibited thrombosis in arterioles and venules. Blockade of GPIb-vWF leads to the inhibition of thrombus formation in the stenosed coronary arteries [84, 85]. In contrast, some autoantibodies to vWF have been shown to increase vWF binding to platelets and activated platelet via binding to the Fc gamma RII receptor. Thus, these autoantibodies may be responsible for a new form of antibody-mediated thrombosis [86].
9.1 Von Willebrand Factor and Thrombotic Thrombocytopenic Purpura (TTP)
Thrombotic thrombocytopenic purpura (TTP) is a disorder of systemic platelet aggregation and a life-threatening microangiopathic syndrome. The underlying pathophysiology of TTP is an extensive presence of platelet and vWF-rich microthrombi. However in hemolytic uremic syndrome (HUS) pathophysiology, microthrombi consist of platelet and fibrin, while in other thrombotic micro-angiopathies (TMAs), varying amounts of infiltrated lymphocytic or neutrophil are interconnected to the microthrombi [87]. Familial TTP/HUS is not common and usually occurs in the immediate postnatal period or infancy, but there are reported cases of delayed onset from the second to third decade of life, too. Nevertheless, more frequently TTP is either idiopathic or secondary to a variety of conditions.
9.1.1 TTP Pathological Mechanisms and Recent Insights
A defect in the protease activity of ADAMTS13 causes the remaining vWF multimers uncleaved in the blood stream, which consequently leads to intravascular thrombosis (Fig. 8).
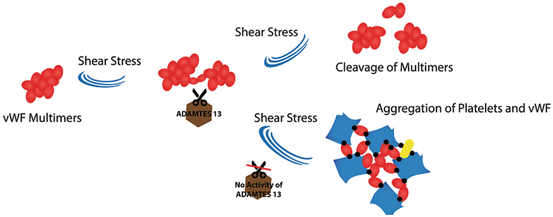
Fig. 8
The role of ADAMTS13 in maintaining intravascular homeostasis: In regular state, when von Willebrand factor multimers are secreted from stimulated endothelial cells, ADAMTS13 enzyme molecules attach to them and then cleave the multimers, but in thrombotic thrombocytopenic purpura (TTP) disease, the absence of ADAMTS13 causes the arrest of multimers cleavage. Thus platelets adhere to the unusually large vWF multimers and cause the risk of developing thrombosis
Evidence has indicated that the aggregating agonist in TTP of all categories might be von Willebrand factor. The data have also shown that uncommon large vWF multimers derived from endothelial cells are the main form of vWF in this process. ULvWF multimers function as the sites for platelet adhesion and aggregation and activate surfaces for the alternative complement pathway. Additionally, activation of complement pathway for generation of terminal complement complexes (C5b-9) occurs in TTP and HUS [88]. Metalloproteinase (ADAMTS13), therefore, plays a vital role in maintaining intravascular homeostasis. It is suggested that a defect in the processing of ULvWFMs after synthesis and secretion by endothelial cells makes patients prone to repeated relapses (Fig. 8). Existing evidence also demonstrated that the metalloproteinase involved in vWF breakdown is not appropriate in children with chronic relapsing TTP. Enzyme deficiency, therefore, might be responsible for congenital chronic relapsing TTP. In contrast autoantibodies might be the main inhibitor for neutralizing vWF metalloproteinase in adults with intermittent or single episode types of TTP. The inhibitors are more frequently IgG subclass in patients with acquired TTP [89], although rare production of IgA and IgM antibodies has been reported. Surprisingly, the rising level of ADAMTS13 inhibitor may be associated with switching the IgG subclasses. This may suggest that cytokine dysregulation may be responsible for the rising inhibitor levels observed in some cases of TTP. However, these types of TTP in adults are likely to be short-term or recurrent autoimmune processes, respectively [89]. Almost one to two-thirds of patients with autoimmune TTP have been reported to show ADAMTS13-specific circulating immune complexes. This may be a pathophysiologic mechanism in this type of TTP, although it has not been methodically investigated [91].
9.1.2 TTP Subgroups
Thrombotic thrombocytopenic purpura (TTP) can be divided into different sub-types; therefore the diagnosis is based on the patient’s history and relevant parameters.
Congenital TTP
The first type of TTP is usually found in neonates with severe jaundice, while the second type is associated with unexplained thrombocytopenia in children and adults. Eventually, the congenital TTP is diagnosed through ADAMTS13 activity <5 %, lack of antibody and the homozygous or heterozygous defects of ADAMTS13 gene.
Drug-Related TTP
Some drugs like quinine and oestrogen-containing medications are associated with TTP. Therefore to prevent relapse, these medications should be avoided and women with a history of TTP should take non-oestrogen containing drugs.
TTP and Pregnancy
TTP may occur frequently in women and therefore can be linked with pregnancy. The greatest risk for development of TTP is around the term and during the post-partum period. Henceforward, the successful outcome of pregnancy in a woman with TTP in early first trimester was achieved after treatment with therapeutic plasma exchange [92].
9.1.3 TTP Diagnosis Based on the Patient’s History and Following Parameters
- 1.
Clinical examination and routine laboratory factors, including blood film review
- 2.
Considering human immunodeficiency virus (HIV), hepatitis B and C viruses and autoantibody screen
- 3.
Detection of ADAMTS13 activity ranks and evaluation of anti-ADAMTS13 antibodies
- 4.
Detection of ADAMTS 13 antigen levels in cases with congenital type of TTP
Management and Treatment
- 1.
Plasma exchange using (PEX) solvent/detergent-treated plasma
- 2.
Exaggeration in frequency and/or volume of PEX procedures in life-threatening cases
- 3.
When two cases of myocardial infarction occur shortly after platelet transfusion, transfusion is contraindicated unless platelet count is low (<20 × 109/L) [93]
- 4.
Intermediate purity Factor VIII infusion
- 5.
Avoiding quinine and oestrogen-containing medications in drug-associated TTP
- 6.
Individualized treatment regimens for congenital TTP according to the patient’s phenotype
- 7.
Treatment considerations for TTP in pregnancy
- 8.
Treatment considerations for TTP in patients with HIV infection
PEX in conjunction with highly active antiretroviral therapy (HAART) (triple or quadruple therapy)
Rituximab in resistant HIV-related TTP
- 9.
Treatment considerations for malignancy-associated thrombotic microangiopathy (PEX should not be used)
- 10.
Additional treatment
Intravenous methylprednisolone or high-dose oral prednisoloneRelated posts:
 Clinical Presentation and Basic Work up in Patients with Suspected Pulmonary Embolism
Prevention of Venous Thromboembolism in Surgical Patients
Clinical Presentation and Basic Work up in Patients with Suspected Pulmonary Embolism
Prevention of Venous Thromboembolism in Surgical Patients
 Diagnosis and Management of Early Deep Vein Thrombosis
Diagnosis and Management of Early Deep Vein Thrombosis
 Thrombosis: Regulatory Mechanisms and Emerging Directions
Thrombosis: Regulatory Mechanisms and Emerging Directions
 Venous Thrombosis
Venous Thrombosis
 Treatment of Venous Thromboembolism in Patients with Cancer
Treatment of Venous Thromboembolism in Patients with Cancer

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


