Clinical
Stage
Absolute lymphocyte count
Lymphocyte doubling time
Bone marrow infiltration pattern
Treatment response
Age
Performance status
Genetic
IGHV mutational status
FISH
Low risk: del(13q)
Intermediate risk: trisomy 12
High risk: del(17p), del(11q), complex karyotype
Mutations
Favorable: MYD88
Unfavorable: TP53, NOTCH1, SF3B1, BIRC3, ATM
Flow cytometry
ZAP-70
CD49d
CD38
Serum
Lactate dehydrogenase
β2-microglobulin
Thymidine kinase
CCL3 and CLL4 serum levels
3 IGHV Mutational Status
B-cell development begins in the bone marrow when hematopoietic progenitor cells (HPCs) differentiate into pro-B cells [54]. These cells undergo sequential rearrangement of the B-cell receptor (BCR) immunoglobulin heavy and light chains, then exit into the peripheral blood as naïve B cells [54]. Upon migration into lymphoid tissue, naïve B cells are activated by antigen-presenting cells and may differentiate into plasma cells or memory B cells [54]. The variable regions of the immunoglobulins, which encode the BCR antigen-binding domains, can acquire somatic mutations that increase the affinity to a specific antigen [54]. The mutational status of clonal immunoglobulin heavy chain variable (IGHV) genes is determined by comparing the IGHV sequence of malignant cells to known germ line sequences [56]. An IGHV is considered mutated if it shares less than 98 % homology to the corresponding germ line sequence [56].
As recognized in the early 1990s, CLL patients can be divided into two groups based on the IGHV mutational status [122]. In about half of the patients, CLL cells express an IGHV gene in germ line configuration, while in the other half, >2 % of the bases are mutated [122]. In 1999, these two groups of patients were reported to have significantly disparate clinical outcomes [34, 64]. Patients with IGHV-unmutated CLL (U-CLL) have a median OS of approximately 9 years compared to 24 years for those with IGHV-mutated CLL (M-CLL) [34, 64]. Furthermore, patients with U-CLL more often have advanced stage disease at diagnosis, progress faster, and require earlier and more frequent treatment [34, 64, 109]. Most cases of transformation to high-grade lymphoma (Richter’s transformation) also occur in U-CLL [86, 126]. Although the incidence of hypogammaglobulinemia is similar in M-CLL and U-CLL, the latter is associated with more infection and infection-related mortality [51]. Unfavorable genetic aberrations, including 17p and 11q deletions, are common in U-CLL, while 13q14 deletion or translocation occur more often in M-CLL [64, 79, 98].
In a subset of patients, the clonal cells express a restricted repertoire of IGHV genes and in some cases, carry BCRs that are structurally virtually identical, referred to as stereotypic BCRs [1, 130]. This suggests that in these cases, CLL originates from a B cell with distinct antigen specificity. In particular, the sequence and length of the complementarity-determining region 3 (CDR3) appear to contribute to stereotypic BCRs and differ between U-CLL and M-CLL [87, 153]. U-CLL cells commonly employ longer, stereotyped CDR3s [130], which are thought to enhance BCR polyreactivity [87]. Indeed, the BCRs of U-CLL tend to be more polyreactive and of lower affinity than those found in M-CLL or normal B cells [69]. Consequently, it has been hypothesized that U-CLL may respond to multiple trophic signals or antigens, leading to more rapid expansion of the malignant clone [131]. In contrast, M-CLL cells appear to be anergic, that is, antigen-hyporesponsive, likely as a result of prior high affinity or repetitive antigen-binding [131]. This may, at least in part, explain the differences in disease pace between the two subtypes. Thus, IGHV mutation status may be viewed as a surrogate of BCR signaling strength (Fig. 1). In addition, chemokines, CCL3 and CLL4, are secreted by activated CLL cells, facilitate crosstalk with the microenvironment, and may serve as markers for BCR signaling [125]. As is the case in an increasing number of B-cell malignancies, antigenic stimulation has emerged as a driver pathway in the pathogenesis of CLL [93, 154].
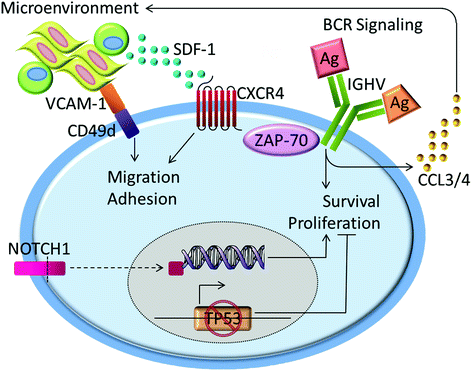
Fig. 1
Key prognostic markers in CLL reflect underlying pathogenic pathways. BCR signaling is a central pathway in the pathogenesis of CLL. The BCR appears to be polyreactive in U-CLL, leading to increased receptor activation and downstream signal transduction. Activated CLL cells secrete chemokines, CCL3 and CLL4, which attract accessory cells to the microenvironment. Migration and adhesion to the microenvironment is further facilitated by ligation of CXCR4 and CD49d on the surface of CLL cells. ZAP-70 appears to directly associate with the BCR to enhance signaling and modulate CXCR4-mediated chemotaxis. Specific genetic lesions promote CLL cell survival and proliferation. Abnormalities in TP53 impair the cellular response to DNA damage and avert apoptosis and growth arrest. Activating mutations in NOTCH1 produce a more stable intracellular signaling protein that regulates the transcription of multiple survival pathways
3.1 Zap-70
Zeta-chain-associated protein of 70 kD (ZAP-70) is a cytoplasmic kinase expressed in T and NK cells [22]. Upon T-cell receptor (TCR) stimulation, ZAP-70 associates with the zeta-chain of TCR and undergoes phosphorylation [22]. ZAP-70 is structurally homologous to spleen tyrosine kinase, which plays an analogous role in BCR signal transduction [22]. Although ZAP-70 is not found in mature peripheral blood B cells, it is expressed in pro- and pre-B cells [32] as well as activated tonsillar and splenic B cells [95]. ZAP-70 is one of the most differentially expressed genes between U-CLL and M-CLL [116, 148]. Because ZAP-70 is highly concordant with IGHV mutational status [31, 97, 108, 116, 148, 155], it has been suggested as a surrogate. ZAP-70 expression appears to be regulated by methylation of the 5′ region of the ZAP-70 gene [28, 30] and heat shock protein 90 chaperoning [21]. The level of expression follows a continuum, remains stable over time and is similar across peripheral blood and bone marrow specimens [38, 108]. Cutoffs ranging from 10 to 20 % of ZAP-70+ CLL cells have been shown to best correlate with IGHV mutational status [31, 97, 108]. ZAP-70 is most commonly detected by flow cytometry, but can also be evaluated by RT-PCR, Western blotting, and immunohistochemistry with comparable results [31, 97, 148]. Nevertheless, it has been difficult to standardize ZAP-70 testing in clinical practice, and the absence of a bimodal distribution results in indeterminate or borderline results for many patients. Recent studies suggest that analysis of ZAP-70 promoter methylation may yield more robust results [29].
Patients with ZAP-70+ CLL have a median survival of 9 years versus 24 years [97], more rapid disease progression [31], and require treatment earlier [38, 108]. ZAP-70+ CLL is associated with advanced clinical stage, higher lymphocyte count, and higher tumor proliferation [38, 113, 127]. Interestingly, patients with ZAP-70+ CLL are more likely to develop autoimmune cytopenias [155]. While ZAP-70 is more often found in U-CLL than M-CLL, it maintains prognostic value even in discordant cases [38, 108].
CLL cells expressing ZAP-70 have an augmented response to BCR activation. ZAP-70 may directly associate with the BCR and enhance downstream signal transduction [24, 25]. Interestingly, this effect is independent of its kinase activity [58]. ZAP-70 also enhances CLL cell migration in response to the chemokines CCL19 and CCL21 [37, 113]. The effect of PI3K inhibition, which impairs survival and adhesion of CLL cells in culture, is attenuated by ZAP-70 expression [80]. In the microenvironment, ZAP-70+ CLL is associated with effector T cells and a cytokine profile that is skewed toward IL-4 and IL-10 [91]. These changes, characteristically seen in advanced stages of disease, are observed even in early stage ZAP-70+ CLL and may contribute to a more aggressive disease course [91].
3.2 Cd49d
CD49d is the α4 subunit of the integrin heterodimers α4β1 and α4β7. α4β1, present on B and T cells, mediates leukocyte development and trafficking through binding to its major ligands, vascular cell adhesion molecule-1 (VCAM-1), and fibronectin [65]. In the B-cell compartment, α4β1 facilitates localization to the bone marrow and germinal centers and promotes survival by up-regulation of anti-apoptotic Bcl-XL [115]. The role of α4β7 is more restricted, directing lymphocytes into intestinal lymphoid tissue via ligation to mucosal addressin cell adhesion molecule-1 [8].
The expression of CD49d is measured by flow cytometry [55, 118, 124]. Tumor cells in approximately 40 % of CLL cases and in a larger proportion of other chronic lymphoproliferative disorders express CD49d [33]. Proposed cutoff values to predict clinical outcome in CLL range from 30 to 45 % [14, 55, 124]. Because the rate of CD49d expression follows a U-shaped distribution across a group of patients, CD49d can be clearly read out as positive or negative in most cases [14, 55].
Patients with CD49d+ CLL have more advanced stages [45], more rapid disease progression, and worse OS [14, 55]. CD49d is associated with a number of unfavorable prognostic factors, including CD38, ZAP-70, and possibly IGHV mutation status [55, 124]. The CD49d gene is hypomethylated in CLL with trisomy 12, leading to increased expression compared to CLL without trisomy 12 [158]. CD49d is superior to Rai stage, ZAP-70, CD38, β2-microglobulin, and del(11q) in prognosticating outcome [14, 124]. In a recent multicenter analysis, CD49d was identified as the only independent flow cytometry-based prognostic factor for OS, along with age, gender, IGHV mutation status, del(17p), and absolute lymphocyte count [14].
In vitro studies have demonstrated the role of CD49d in transendothelial migration of CLL cells [137] and recruitment of monocytes, endothelial, and stromal cells to the bone marrow microenvironment [157]. Furthermore, α4β1, together with CD44v, anchors matrix metalloproteinase-9 to the surface of CLL cells [112]. As a result, malignant cells are arrested in the bone marrow and lymph node, thereby presumably enhancing the nurturing effect of the microenvironment on the tumor cells [112]. CD49d may also competitively engage ligands in the bone marrow and displace HPCs, leading to increased numbers of HPCs in the circulation [118]. In addition, CD49d ligation leads to up-regulation of anti-apoptotic Bcl-2 and Bcl-XL and down-regulation of pro-apoptotic Bax, thus preventing spontaneous [35, 81] and fludarabine-induced apoptosis [36] of CLL cells in culture.
3.3 Genetic Aberrations
Interphase cytogenetic analysis using fluorescent DNA probes for in situ hybridization (FISH) allows detection of chromosomal abnormalities in over 80 % of non-dividing CLL cells [39]. Previously, CLL cells required mitogen stimulation for traditional chromosome analysis in metaphase, which yielded an abnormal karyotype in only half of patients [73]. Using FISH, the most commonly observed abnormality is 13q deletion in over half of patients, followed in order by 11q deletion, 12q trisomy, and 17p deletion [39]. These can present as a sole abnormality or in combination with one another [39].
Survival of early stage patients ranges from a median of 133 months with 13q deletion to 79 months with 11q deletion and 32 months with 17p deletion [39]. Deletions in 11q and 17p occur almost exclusively in U-CLL and have independent prognostic value [79]. These deletions are also associated with advanced disease and rapid disease progression requiring early treatment [39]. In contrast, one-third of patients with 13q deletion do not require any therapy [39]. The prognosis of del(17p) CLL has been correlated with the percentage of affected CLL cells [136]. In the largest series reported, patients with del(17p) in ≤25 % of CLL cells had a relatively indolent disease course and a 3-year OS of ≥90 % [136]. Del(17p) occurring in M-CLL also appears to be compatible with relatively slower disease progression [57].
Genetic mutations and chromosomal abnormalities have significant overlap in CLL. Loss of tumor suppressor genes contributes to the aggressive nature of CLL with unfavorable karyotype. Mutation in TP53, located on chromosome 17p, is found in approximately 80 % of del(17p) CLL [60, 156]. The combination of TP53 mutation and del(17p) predicts shorter progression-free survival (PFS) than del(17p) alone [60]. In addition, sole TP53 mutation, without del(17p), portends a similar prognosis to del(17p) CLL [156]. The ataxia-telangiectasia (ATM) gene on chromosome 11q is mutated in 30 % of del(11q) CLL [4]. Mutation in ATM, which coordinates the cellular response to DNA damage, is associated with shorter OS and PFS [4]. Conversely, the favorable outcome observed in del(13q) CLL, which is superior to that of normal karyotype, has not been well elucidated. Down-regulation of miR15 and miR16 appears to play a role [20], leading to an increase in target proteins, including anti-apoptotic Bcl-2 [27].
Whole genome and exome sequencing has identified a number of recurrent genetic mutations with prognostic relevance. NOTCH1 is a commonly mutated gene in CLL [104] and partially correlates with trisomy 12 [7, 83]. Patients with NOTCH1-mutated CLL have shorter survival and increased risk of Richter’s transformation [104]. Mutation in the spliceosomal gene, SF3B1, is associated with more rapid disease progression. In contrast, mutations in MYD88, which encodes an adaptor protein in interleukin-1/toll-like receptor signaling, tend to be more common in younger patients [104] with mutated IGHV genes [143] and have relatively favorable disease course [88]. Importantly, it appears that specific genetic abnormalities, including trisomy 12, del(13q), and MYD88 mutation, appear to represent driver events in CLL, while other aberrations, such as del(17p) and mutations in TP53, SF3B1, and ATM, occur later with disease progression and increase in frequency in patients with relapsed and refractory disease [82].
4 First-Line Treatment
4.1 Standard Chemoimmunotherapy
Treatment of CLL is generally reserved for patients with active disease or constitutional symptoms fulfilling specific treatment criteria as outlined by the International Workshop on Chronic Lymphocytic Leukemia (Table 2) [62]. The purine analogue, fludarabine, and its combinations have been the mainstay of therapy for many years. In a landmark trial led by the Cancer and Leukemia Group B (CALGB), initial therapy with fludarabine, compared to chlorambucil, improved overall response rate (ORR) from 37 to 63 % [106]. However, perhaps owing to its crossover design, no difference in OS was initially found [106]. Subsequently, a long-term follow-up analysis suggested an eventual benefit in OS for patients treated with fludarabine [107]. However, the benefits of fludarabine appear to be limited to younger patients. The German Chronic Lymphocytic Leukemia Study Group (GCLLSG) randomized older patients with a median age of 70 years to first-line therapy with fludarabine or chlorambucil [44]. While fludarabine improved ORR, it did not change OS or PFS [44]. Rather, patients treated with chlorambucil had a median OS of 64 months compared to 46 months with fludarabine, although this difference was not statistically significant [44]. A retrospective analysis of consecutive trials from CALGB also found similar OS and PFS between fludarabine and chlorambucil treatment in the elderly [151].
Indications for treatment | Criteria (at least one of the following) |
|---|---|
Marrow failure | Anemia (hemoglobin <10 g/dL) |
Thrombocytopenia (platelet count <100 K/μL) | |
Splenomegaly | Spleen ≥6 cm below left costal margin |
Progressive/symptomatic | |
Lymphadenopathy | Lymph node ≥10 cm in diameter |
Progressive/symptomatic | |
Lymphocytosis | Lymphocyte doubling time <6 months1 |
Lymphocyte count increase of >50 % in 2 months1 | |
1Should not be used as only criteria if initial lymphocyte count <30 K/μL | |
1Exclude infection as cause for worsening lymphocytosis | |
Autoimmune anemia or thrombocytopenia | Poor response to corticosteroids or other standard therapies |
Constitutional symptoms | Weight loss ≥10 % in 6 months |
Fatigue (ECOG PS ≥2) | |
Fever >38 °C for ≥2 weeks2 | |
Night sweats >1 month2 | |
2Exclude infection as cause for fever or night sweats |
For younger, physically fit patients, the development of fludarabine-based combination regimens has led to a new standard in CLL treatment. The addition of cyclophosphamide to fludarabine (FC) increased complete response (CR) rate from 5 to 20 % and median PFS from approximately 20–40 months in two prospective, randomized trials of 375 patients from Germany [43] and 278 patients from the United States [49]. However, neither trial found a difference in OS [43, 49]. Meanwhile, rituximab monotherapy was demonstrated to have moderate activity against CLL [61, 96]. In addition, several phase II trials supported the combined use of rituximab and fludarabine, with or without cyclophosphamide [18, 76, 123, 135, 144]. Ultimately, the GCLLSG conducted a prospective, randomized, multicenter study (CLL8) to examine the benefit of adding rituximab to FC (FCR) in 817 treatment-naïve CLL patients [63]. Chemoimmunotherapy improved OS at 3 years from 83 to 87 %, ORR from 80 to 90 %, CR rate from 22 to 44 %, and median PFS from 32.8 to 51.8 months [63].
4.2 Options for Older Patients
Bendamustine with rituximab (BR) may be better tolerated in older patients with comorbidities. A phase II study of 117 previously untreated patients demonstrated an ORR of 88 %, a CR rate of 23 %, median event-free survival of 33.9 months, and OS of 90.5 % at a median follow-up of 27 months [48]. A quarter of these patients were older than 70 years and 35 % had impaired renal function [48]. Toxicities were primarily hematologic and included grade 3 or 4 neutropenia in 19.7 % of patients [48], compared to 34 % with FCR [63]. Subsequently, the GCLLSG led an international phase III trial randomizing 688 patients to FCR or BR [47]. Results of the interim analysis were presented at the American Society of Hematology 2013 annual meeting [47]. Although there was no difference in OS, FCR produced a higher CR rate (47.4 % vs. 38.1 %) and longer PFS (85 % vs. 78.2 %) at 2 years, but only in patients younger than 65 years [47]. These benefits were offset by an increased risk of severe neutropenia and infection [47].
Obinutuzumab, a humanized, type 2, glycoengineered antibody against CD20, was recently demonstrated to improve response and survival when added to chlorambucil [59]. The GCLLSG randomized 781 patients with a median age of 73 years and multiple co-morbidities to chlorambucil with or without obinutuzumab (G-Cbl) or rituximab (R-Cbl) [59]. At a median follow-up of 14 months, G-Cbl reduced the risk of death from 20 to 9 % compared to patients treated with chlorambucil alone [59]. G-Cbl was also superior to R-Cbl with respect to response (ORR 65.1 % vs. 78.4 %; CR rate 7 % vs. 20.7 %), disease progression (median PFS 26.7 vs. 15.2 months), and survival (risk of death 8 % vs. 12 %) [59]. However, the dosing of obinutuzumab was reasonably different from that of rituximab [59] so as to make a direct comparison between the two antibodies difficult.
5 Relapsed/Refractory CLL
Definitions of relapsed/refractory disease facilitate clinical decision making and scientific study [62]. Relapse occurs in patients previously in complete or partial remission who progress after 6 or more months [62]. Refractory disease is defined by treatment failure or disease progression within 6 months of treatment [62]. Patients refractory to purine analogue-based regimens are considered particularly high risk [62]. Although the biology of non-responders is conceivably different from those who relapse after a period of disease control, most clinical trials consider relapsed or refractory disease as one entity.
Treatment options for relapsed/refractory CLL are growing despite the fact that a standard treatment has yet to be established (Table 3). Consequently, participation in clinical trials should be considered for the majority of patients in this setting. For those who previously achieved a durable remission, repeating the same regimen may be efficacious at relapse. In particular for patients with early relapse or refractory disease, novel agents (discussed below) have demonstrated significant activity.
Table 3
FDA-approved drugs for CLL
Drug | Mechanism | Approval | Indication |
|---|---|---|---|
Chlorambucil | Alkylating agent | 1957 | Unspecified |
Cyclophosphamide | Alkylating agent | 1959 | Unspecified |
Fludarabine | Nucleotide analogue | 1991 | Relapsed/refractory |
Alemtuzumab | Monoclonal antibody | 2001 | Relapsed/refractory |
2007 | First-line | ||
Bendamustine | Alkylating agent | 2008 | Unspecified |
Ofatumumab | Monoclonal antibody | 2009 | Relapsed/refractory |
2014 | First-line in combination with chlorambucil | ||
Rituximab | Monoclonal antibody | 2010 | First-line in combination with fludarabine and cyclophosphamide for CD20+ CLL |
Obinutuzumab | Monoclonal antibody | 2013 | First-line in combination with chlorambucil |
Ibrutinib | Kinase inhibitor | 2014 | Relapsed/refractory |
5.1 Chemoimmunotherapy
FCR induced an ORR of 74 % and a CR rate of 30 % in 284 patients with relapsed/refractory CLL enrolled on an open-label, phase II trial [5]. Response was preserved in patients previously treated with fludarabine or rituximab, but not in those who received a prior alkylating agent [5]. As expected, patients who responded to fludarabine in the past had a better ORR with FCR salvage [144]. Median PFS and OS were 21 and 47 months, respectively [5].
5.2 Monoclonal Antibodies
Approximately half of patients respond to ofatumumab in the salvage setting [146], including those previously treated with rituximab [147]. Median PFS and OS are 5.5 and 13–16 months, respectively [146, 147].
Alemtuzumab, a humanized monoclonal antibody against CD52, has been studied both as first-line and salvage treatment in CLL. Relapsed/refractory patients treated with alemtuzumab have an ORR of 33–42 %, median PFS of 8–12 months, and median OS of 16–19 months [75, 100, 132]. Response is significantly higher in treatment-naïve patients [85]. However, alemtuzumab also causes prolonged lymphopenia, increasing the risk of opportunistic infections (OI) [75, 85, 100, 132]. CMV reactivation is the most common OI, occurring in up to 30 % of patients receiving alemtuzumab [75, 85, 132]. Grade 3 or 4 CMV infections have been reported, but respond to ganciclovir [132]. Accordingly, regular monitoring and prompt treatment of CMV viremia are strongly encouraged. In 2012, alemtuzumab or Campath® was withdrawn from the US and EU markets in anticipation of a relaunch under a different trade name Lemtrada® for the treatment of multiple sclerosis. Although Lemtrada® has not been approved in the USA, alemtuzumab remains available through the Campath® distribution program.
5.3 Kinase Inhibitors
Much interest has been focused on targets of the BCR signaling pathway. Ibrutinib, an inhibitor of Bruton tyrosine kinase, was recently approved for use in previously treated CLL [19]. In a landmark trial, patients treated with ibrutinib had an ORR of 71 %, PFS of 74 %, and OS of 83 % at 26 months, as well as a lower risk of severe hematologic toxicity and infection than conventional salvage regimens [19]. An international, phase 3 trial comparing ibrutinib to ofatumumab in 391 patients found an ORR of 43 % versus 4 % and a 78 % reduction in risk of progression or death with ibrutinib [17]. Notably, one-third of these patients had deletion of 17p [17].
Idelalisib inhibits PI3Kδ and results in an ORR of 81 % when combined with rituximab in the salvage setting [53]. Compared to rituximab monotherapy, the addition of idelalisib improves PFS from 46 to 93 % and OS from 80 to 92 % at 12 months [53]. These kinase inhibitors cause transient lymphocytosis [19, 53] comprised of biologically inert cells [67, 152], that egress from lymph nodes and spleen [68].
5.4 BH3 Mimetics
BH3 mimetics are small molecule inhibitors of the Bcl-2 family of proteins that regulate cell survival and death. Navitoclax, a non-specific inhibitor of Bcl-2, Bcl-XL, and Bcl-W, was studied in 29 patients with relapsed/refractory CLL with an ORR of 31 %, including cases with 17p deletion. However, it also uniformly causes dose-limiting thrombocytopenia as a result of Bcl-XL inhibition. Consequently, ABT-199, a BH3 mimetic specifically targeting Bcl-2, was developed and shown to have preclinical activity against CLL [134, 142], multiple myeloma [138], AML [10, 94], and breast cancer [141]. Several phase I trials evaluating ABT-199, as monotherapy or in combination with monoclonal antibody or bendamustine, are currently underway.
5.5 Lenalidomide
Immunomodulating agent, lenalidomide, was initially studied as salvage monotherapy with an ORR of 32–47 % [23, 46]. Response is improved to 66 % when combined with rituximab and is associated with an OS of 71 % at 3 years [6]. Lenalidomide may cause grade 3 or higher neutropenia in up to 70 % of patients [6, 23] and is often associated with a cytokine release syndrome and tumor flare reactions [3, 23, 46]. In 2013, a phase 3 trial comparing lenalidomide to chlorambucil in previously untreated patients older than 65 years was terminated by the US Food and Drug Administration due to increased deaths in the lenalidomide arm [140]. Thirty-four of 210 patients treated with lenalidomide and 18 of 211 patients treated with chlorambucil died [140]. The Chronic Lymphocytic Leukemia Research Consortium subsequently reported the results of a phase 2 trial of initial therapy with lenalidomide and rituximab [72]. Among 29 patients older than 65 years, the ORR was 79 %, median PFS was 20 months, and 4 had died over a median follow-up of 20 months [72].
5.6 Cellular Therapy
Allogeneic hematopoietic stem cell transplantation (HSCT) provides graft-versus-leukemia effect and a potential for cure to patients with high-risk CLL [40]. Myeloablative conditioning followed by matched related [90] or unrelated [102] HSCT can lead to durable remissions, but carries a 38–46 % risk of treatment-related mortality (TRM). With reduced-intensity conditioning, TRM is reduced to 15–22 %, improving OS without sacrificing response [120, 128, 129]. However, graft-versus-host disease (GVHD) remains a significant problem, with grade 2–4 acute and chronic GVHD in 55 and 50–75 % of patients, respectively [120, 128, 129].
Genetically modified T cells expressing chimeric antigen receptor (CAR) have specific activity against their ligands. The receptor is engineered to directly activate T cells upon ligation without major histocompatibility complex restriction. In phase I trials, patients infused with autologous anti-CD19 CAR T cells were able to achieve stabilization of disease, partial remission (PR), or in some instances, sustained CR [13, 74, 103]. A subset of CAR T cells acquires a central memory phenotype in vivo, allowing for persistent activity against CLL cells [74]. Allogeneic CAR T cells have been studied in allogeneic HSCT recipients at relapse [78]. Infusion of CAR T cells from the original donor led to an ORR of 20 % and stable disease in 8 of 10 patients [78].
6 Predictive Markers
Whereas prognostic markers inform the overall clinical course of a disease, predictive markers identify patients who are susceptible to specific therapies. Ideally, predictive markers can help physicians tailor treatment for individual patients, thereby avoiding wasted time and toxicities from less-effective options. Despite the apparent wealth of prognostic markers in CLL, the literature on predictive markers is limited.
6.1 Deletion of 17p
Deletion of 17p or mutation in TP53, one of the best established predictive markers, is typified by poor response to standard chemoimmunotherapy. Among 51 patients with del(17p) enrolled in the CLL8 trial comparing FC to FCR, only 1 patient achieved CR compared with a CR rate of 44 % for all patients following treatment with FCR [63]. At 3 years, 19 patients with del(17p) were alive and all had progressive disease [63]. A follow-up report showed comparably poor outcomes in CLL with mutated TP53; the majority of which were associated with loss of 17p [133]. As expected, responses are worse when FCR is given as salvage therapy [5]. Furthermore, patients with del(17p) or TP53 mutations also appear resistant to combinations of bendamustine or chlorambucil with rituximab [48, 50]. In contrast, small molecule inhibitors of BCR signaling, such as ibrutinib [19] or idelalisib [53], have shown promising activity against this high-risk subgroup. Alemtuzumab is also effective for both treatment-naïve [26, 70] and relapsed/refractory patients [84, 101, 132] irrespective of 17p or TP53 status.
The European Group for Blood and Marrow Transplantation (EBMT) considers 17p deletion as an indication for allogeneic HSCT [40]. A retrospective analysis of del(17p) CLL from the EBMT database found an ORR of 84 % in 19 patients with stable or progressive disease and an ORR of 94 % in 17 patients in PR at the time of transplantation [121]. Importantly, 16 patients remained in CR after a median follow-up of 39 months [121]. In a phase 2 trial of reduced-intensity conditioning with allogeneic HSCT (CLL3X), event-free survival and OS were similar between patients with and without 17p deletion [41]. Furthermore, long-term remission with MRD negativity was achieved in 6 of 13 cases of del(17p) CLL [41, 42].
6.2 Deletion of 11q
As previously mentioned, 11q deletion is another adverse prognostic marker associated with a median OS of 49 months [39]. Unlike del(17p), patients with del(11q) CLL respond well to FCR, achieving an ORR of 93 %, CR rate of 51 %, 3-year PFS of 64 %, and 3-year OS of 94 % in the CLL8 trial [63]. These response and survival rates were similar to those observed in patients without del(17p) or del(11q) [63]. A retrospective analysis of 69 patients with del(11q) CLL corroborated the benefits of FCR-based regimens in this genetic subgroup [139]. Without an alkylating agent, however, outcomes are significantly worse for del(11q) CLL, with a median PFS and OS of 25 and 63 months, respectively [150]. Therefore, alkylating agents appear to be particularly valuable in reversing the poor prognosis of del(11q) CLL.
6.3 Novel Mutations
The role of novel genetic mutations as predictive markers has recently been explored by several groups. Methods include targeted and whole-exome sequencing (WES) of cases drawn from both retrospective series and prospective cohorts. WES of fludarabine-refractory patients has identified mutations in TP53, NOTCH1, SF3B1, or BIRC3 in 70 % of cases [89].
Mutations in NOTCH1 occur in 20 % of fludarabine-refractory disease [117]. NOTCH1-mutated patients treated with chlorambucil or a fludarabine-based regimen have shorter survival and more rapid disease progression compared to wild-type cases [99]. Median OS after treatment may approach that of TP53-mutated CLL as demonstrated by Rossi et al. [117]. Interestingly, NOTCH1 mutation identifies a unique subgroup of patients who appear to have no benefit from the addition of rituximab to FC [133].
6.4 Minimal Residual Disease
Minimal residual disease (MRD), a sensitive measure of tumor burden post-treatment, predicts shorter PFS [11, 41, 48, 92, 114] and OS [110, 119] for most types of therapy, including standard chemoimmunotherapy and HSCT. Flow cytometry or molecular assays reliably detect residual tumor at a frequency of >1 in 10,000 cells [62]. In addition to evaluating for treatment response, MRD status can be serially monitored to track disease activity [92, 110, 114]. For the most part, conversion from a negative to positive MRD state heralds clinical relapse [92, 110, 114]. Although a powerful predictor of duration of response, MRD status can only be determined after therapy. A practical application may be to stratify patients for additional therapy based on MRD status. For example, patients achieving MRD-negative CR during induction therapy may be adequately treated with an abbreviated course, while those with MRD positivity at the completion of treatment may benefit from consolidation or maintenance. A comprehensive review on this topic was recently published by Böttcher et al. [12].
Related posts:
 Hematopoietic Cell Transplantation in Non-Hodgkin’s Lymphomas
Hematopoietic Cell Transplantation in Non-Hodgkin’s Lymphomas
 Lymphoproliferative Disorders
Lymphoproliferative Disorders
 of Non-Hodgkin Lymphomas: Diagnosis and Response-Adapted Strategies
of Non-Hodgkin Lymphomas: Diagnosis and Response-Adapted Strategies
 Expression Profiling in Non-Hodgkin Lymphomas
Expression Profiling in Non-Hodgkin Lymphomas
 Therapeutic Strategies and New Treatment Paradigms for Follicular Lymphoma
Therapeutic Strategies and New Treatment Paradigms for Follicular Lymphoma
 of T-Cell Lymphomas: Diagnosis and Biomarker Discovery
of T-Cell Lymphomas: Diagnosis and Biomarker Discovery
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


