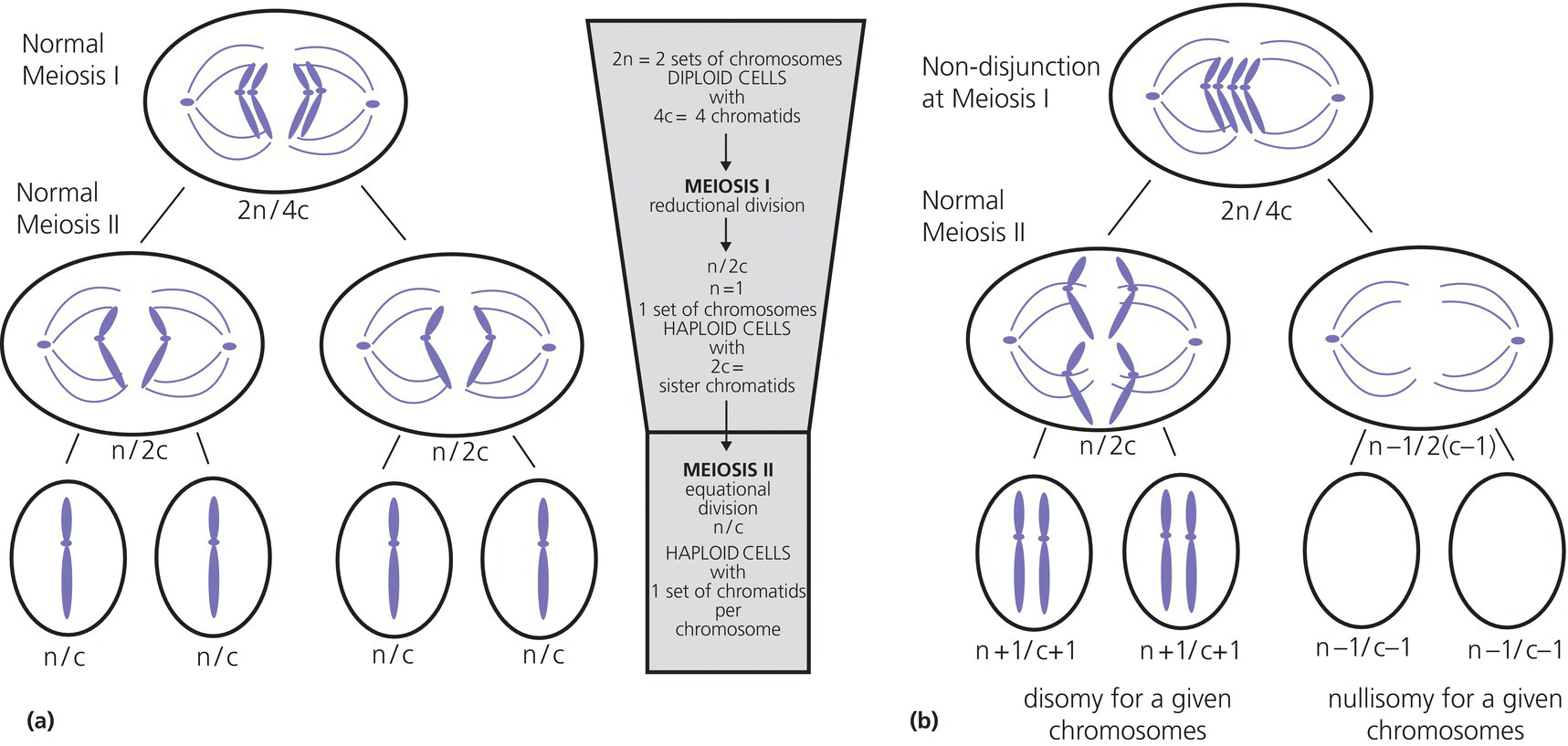
13 Comparative genomic hybridization (CGH) and next‐generation sequencing (NGS) have revolutionized genetics and molecular endocrinology. The use of these new technologies is spreading; clinicians nowadays need to understand the basic concepts and techniques of genetics in order to explain them to patients to obtain informed consent. Moreover, NGS delivers such a wealth of data that geneticists also need the help of clinicians to filter results and to find causative mutations related to the phenotype. Therefore, clinicians need an understanding of genetics and an awareness of these new techniques, their invaluable advantages, as well as their inherent limitations. A complete list of definitions is provided in Table 13.1. Some concepts useful for understanding the NGS technologies are explained in more detail hereafter. Table 13.1 Definition of basic concepts in molecular genetics. A protein‐coding gene is a linear sequence of nucleotides (a segment of deoxyribonucleic acid, DNA) that provides coded instruction for ribonucleic acid (RNA) synthesis and translation to protein. The exons are the coding sequences of a gene, which are interrupted by non‐coding sequences, the introns. The human genome contains 3 billion nucleotides or 3080 million base pairs (Mb) with about 26 000 genes which contain 234 000 exons and 208 000 introns (Scherer 2008). On average, there are 8.8 exons and 7.8 introns per gene (Sakharkar et al. 2004). The totality of all exons of the genome is defined as the exome. Although the exome covers 1–2% of all the genome, approximately 85% of the disease‐causing mutations arise within these protein‐coding regions. The genome is the complete sequence of DNA in an organism. More than 98% of the genome does not code for protein, but it does contain regulatory regions and also several transcribed RNAs (such as long non‐coding RNAs), many of which play roles in the regulation of gene function. The genome is organized and divided into chromosomes, which are structures within the cells that contain the genetic material (Brooker 2018). The term chromosome means ‘coloured body’ which refers to the appearance of chromosomes after coloration through dyes. Human cells are diploid, which means that they have two sets of 23 chromosomes. The first 22 chromosomes are numbered according to their size: chromosome 1 is the largest with 250 Mb and chromosomes 21 and 22 only ~50 Mb. The X chromosome is 155 Mb long and the Y, 58 Mb. Gene density is not equal across the chromosomes, e.g. chromosome 19 contains 26 genes/Mb and the Y chromosome only 3.5 genes/Mb. Human cells are diploid. They contain two sets of chromosomes, 23 pairs of chromosomes for a total of 46 chromosomes (Figure 13.1). In contrast, gametes (sperms and egg cells or their precursors) are haploid, which means that gametes contain a total of 23 chromosomes, i.e. half that of normal diploid cells. To produce gametes, normal cells have to reduce the number of their chromosomes by meiosis, the term given to cell division with a reduction of the genetic material. Meiosis is a succession of two cell divisions which reduce the set of chromosomes from 2 to 1. It is during the first cell division (meiosis I) that crossing‐over of the genetic material occurs, followed by disjunction, which is the division of the genetic material through the centrosomes. Non‐disjunction refers to a failure of the chromosome set to separate correctly. Non‐disjunction in meiosis I is the major source of abnormality in the number of chromosomes in a cell (aneuploidy, see section ‘Aneuploidy’), but non‐disjunction can also occur in meiosis 2. Figure 13.1 (a) schematic view of a normal meiosis for one chromosome pair, each replicated into chromatids (4c) with first division into two cells each containing two sister chromatids (2c) then second division resulting in four cells each containing one set of chromatids (c), also called a monad equivalent to a haploid chromosome. (b) Example of a non‐disjunction in meiosis I results in an aberrant number of chromosomes in the gametes so that after the second division, two cells are disomic for the chromosome and two cells are nullisomic. Aneuploid cells have an abnormal number of chromosomes, and aneuploidy is often associated with human syndromes such as Down’s, Klinefelter, or Turner syndromes. Aneuploidy may arise from several cytogenic mechanisms, but in humans meiotic or mitotic non‐disjunction is the usual mechanism. Non‐disjunction in maternal meiosis 1 is the most common cause of autosomal trisomy and is associated with maternal age (see Figure 13.1). Of note, aneuploidies are genetic conditions that are often de novo (not inherited): thus, while the majority of inherited conditions are genetic, not all genetic conditions are inherited. Even if obvious to geneticists and physicians, this latter point is crucial to explain when counselling parents of children presenting with aneuploidy. The International System for Human Cytogenetic Nomenclature (ISCN) is an official standard for human chromosomes. Normal or abnormal chromosome composition in humans is designated as follows: The normal female chromosomes is thus designated 46,XX and the normal male chromosome is designated 46,XY. An abnormal number of chromosomes is designated first by the total number of chromosomes, the appropriate number of sex chromosomes, and the additional or missing chromosome is identified by + or −, followed by the specific responsible chromosome. Thus, a male with trisomy 21 is designated 47,XY,+21. Examples are listed in Table 13.2 and further complementary definitions and explanations of isochromosomes and ring chromosomes are provided in Figure 13.2. For more complex cases, we refer to this link of the Human Genome Variation Society (HGVS): http://www.hgvs.org/mutnomen/ISCN.html. Table 13.2 Example of chromosome nomenclature following ISCN standard. Figure 13.2 (a) The parts of a chromosome: the centromere divides the chromosome into a short arm (abbreviated p) and a long arm (abbreviated q). Normal or abnormal chromosomes can be classified relative to the position of the centromere: metacentric chromosomes with p and q of equal length; submetacentric chromosomes with q slightly greater than p; acrocentric chromosomes with q much greater than p, with a nearly terminal centromere, and telocentric chromosomes with a terminal centromere. (b) Isochromosome (drawn here in metaphase): a chromosome with identical arms. This abnormal chromosome arises when the centromere divides horizontally rather vertically; the telocentric product then replicates to produce a metacentric chromosome. (c) Isodicentric chromosome (drawn here in metaphase): a chromosome with two centromeres arising when breakage occurs in the chromatid arms. (d) Ring chromosome: arising when breakages occur in the telomeric region of the both arms and the telomeres then stick together to form a ring. Mutations may be defined as any change in DNA sequence (substitutions, additions, or deletions), which may be inherited or which arise de novo. This definition raises some problems: the difference between mutations and variants may be difficult to determine. To link a variant (a genotype) to a range of signs and symptoms (a phenotype), a strong association should be made between the presence of the variant and the disease. This is referred to as co‐segregation of the genotype with the phenotype. The different types of mutations are presented in Table 13.3. Of note, germline mutations arise in cells destined to develop into gametes and can be transmitted to offspring while somatic mutations occur in single cells in developing somatic tissue and are not transmitted. Table 13.3 Different types of mutations. The HGVS provides recommendations for nomenclature of variants (den Dunnen et al. 2016). All mutations should be described at the most basic level, the DNA level, and in relation to an accepted reference sequence. The reference sequence should be public and clearly described. If the mutation is described at the genomic level, the assembly of the genome used should be specified (e.g. currently the reference genome is hg38). If the mutation is described at the level of coding DNA, the isoform should be specified, e.g. NM_014080.4 is the reference coding sequence for the dual oxidase 2 (DUOX2) gene. A letter prefix should be used to indicate the type of reference sequence used. The accepted prefixes are: Descriptions at the DNA, RNA, and protein levels are different.
An Endocrinologist’s Guide to Genetics in the Age of Genomics
Introduction
Basic concepts
Term
Definition
Base
Nucleic acid. There are five different bases subdivided into two categories, the purine bases, adenosine (A) and guanine (G), containing a double‐ring structure; and the pyrimidine bases thymine (T), cytosine (C) and uracil (U), containing a single‐ring structure.
DNA
Deoxyribonucleic acid is a nucleoside which contains a base (A, G, T, or C) linked to a sugar backbone, deoxyribose.
RNA
Ribonucleic acid is a nucleoside which contains a base (A, C, U, or C) linked to a sugar backbone, ribose.
Nucleotide
Nucleosides (DNA or RNA) link to one or more phosphate groups (e.g. ADP, ATP) to form nucleotides. Nucleotides are linked together linearly to form a strand of DNA or RNA. Two strands of DNA interact to form a double helix.
Chromosomes
Structures in the living cell that contain genetic material in the form of a double helix folded with proteins that influence its 3D structure and activity.
Chromatid
After replication, the two copies of a chromosome are called chromatids. There are joined by a centromere to form a unit known as sister chromatids or dyad. After cell division, a chromosome contains a set two chromatids (the long and the short arms of the chromosome) or a monad. Consequently, a normal human cell in interphase has 46 chromosomes and 92 chromatids.
Genome
The DNA found in all the chromosomes.
Gene
A single unit of genetic information. Protein‐coding genes express their information through transcription (RNA expression from DNA), followed by translation (protein synthesis from copy RNA). The pathway of expression from DNA to RNA to protein is called the central dogma of genetics.
Exons
The protein‐coding sequence within a gene.
Introns
The non‐coding sequence within a gene, located between exons. Introns are removed during transcription through a process called splicing.
Diploidy
The existence of pairs of chromosomes (2n) in human cells.
Haploidy
The existence of one set of chromosomes (n) in gametes.
Aneuploidy
An odd number of chromosomes (2n +/− n).
Genes, exome, and genome
Chromosomes
Gametogenesis and meiosis
Aneuploidy
Cytogenetic nomenclature
Official designation
Description
46,XY
Normal male karyotype
46,XX
Normal female karyotype
47,XXY
Klinefelter syndrome
48,XXYY
Variant of Klinefelter syndrome
45,X
Monosomy X; Turner syndrome
45,X/46,XX
45,X/46,XX mosaicism (Turner mosaicism)
46,X,i(Xq)
A rare type of Turner syndrome with Xp monosomy and Xq trisomy due to an Xq isochromosome
46,X,r(X)
Female with a ring X chromosome
47,XX,+21
Female with trisomy 21
46,X,del(X)(p21)
Terminal deletion of the short arm of X distal to band 21. 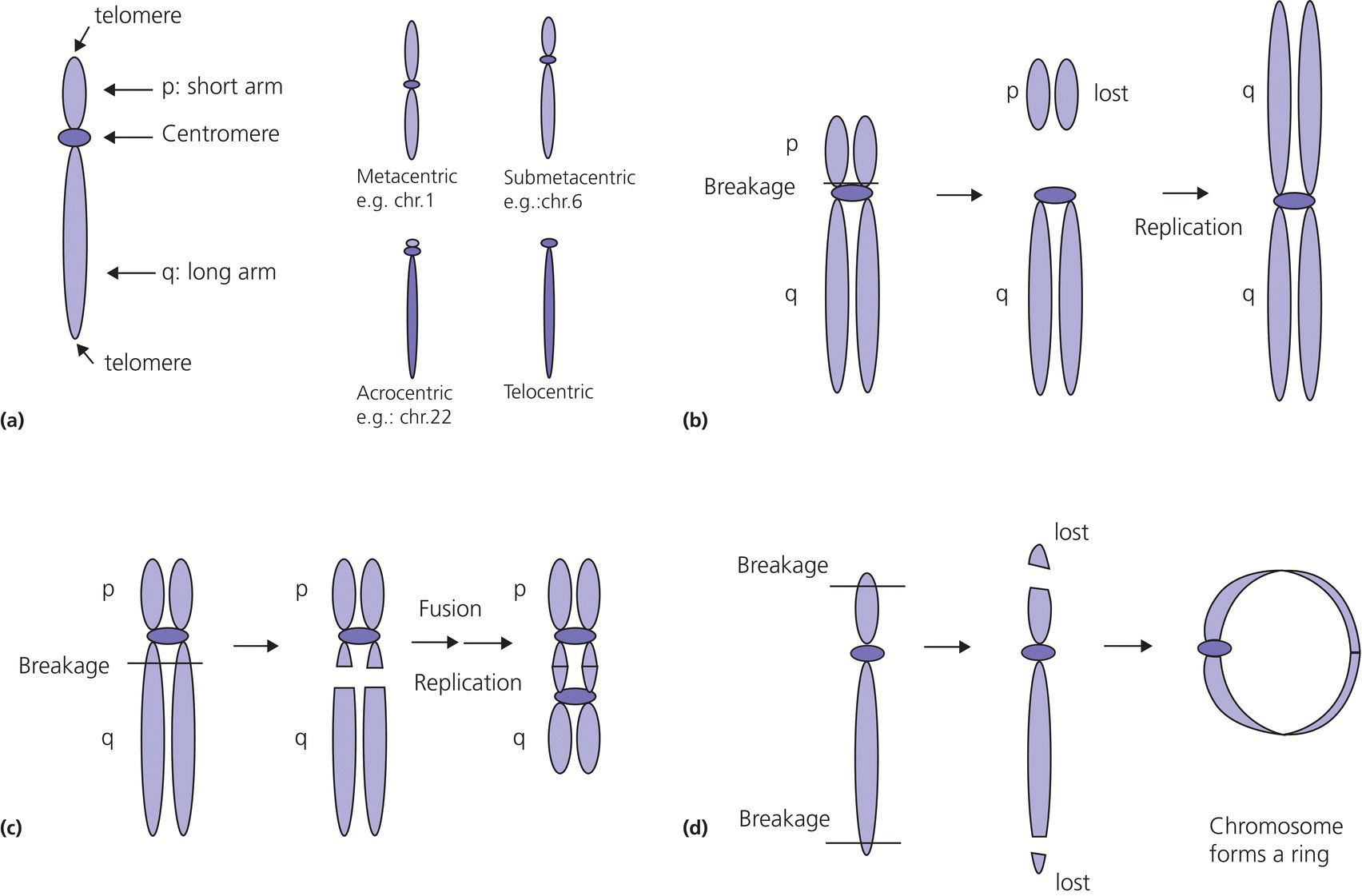
Mutations and mode of inheritance
Mutations
Type
Effect
Example
Silent mutations
No change of amino acid
CAC → CAT (His to His)
Missense mutations
Change of amino acid
AGC → ATC (Ser to Ile)
Nonsense mutations
Replacement of an amino acid codon by a stop codon
TGT → TGA (Cys to STOP)
Splice site mutations
Create or destroy signals for exon/intron splicing
CTGgtaag → CTGatag (247 + 1G → A)
Frameshift mutations (deletions/insertions)
Triplet codon is read wrongly
Gene deletion
Complete gene is missing
Protein is missing
Mutation nomenclature
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


