Introduction
Throughout history, humans have sought to understand the differences between biological sexes and the mechanisms responsible for these biological differences. Normal gonadal differentiation and sex development depend on the meticulous choreography and synchrony of a network of endocrine, paracrine, and autocrine signaling pathways. This network involves the actions and interactions of specific genes, epigenetic influences, transcription factors, and hormones. Perturbations of the intricate network of gene regulation and gene expression governing fetal gonadal development result in disorders of sex development (DSD). Approximately one in 4000 infants are estimated to be born with a DSD. However, this estimate increases if all variations of sex development including cryptorchidism and all forms of hypospadias are included. The prevalence for some subgroups have been merely accurately reported, such as 46,XY females at 6.4/100,000; 4.1/100,000 for androgen insensitivity syndrome (AIS); and 1.5/100,00 for gonadal dysgenesis.
DSDs comprise a spectrum of disorders in which chromosomal, genetic, gonadal, hormonal, or anatomic aspects of sex are atypical. Understanding the developmental biology and embryology of the urogenital system is crucial to categorizing and identifying the molecular basis of the disorder in an individual patient. It has become apparent that multiple genes, epigenetic factors, environmental factors, and protein interactions are involved in the processes of gonadal differentiation and sex development. The pediatric endocrinologists’ role in the care of an infant with ambiguous genitalia begins at the time of initial presentation—birth, childhood, adolescence, or before birth for infants identified through prenatal testing. The physician’s responsibility for the management of this child starts early and continues until transition to adult care providers is deemed appropriate.
Following the 2006 Consensus Statement regarding the management of intersex disorders, knowledge regarding the molecular basis of sex differentiation/development has expanded. The number and capability of the genetic tools available to identify the molecular basis have increased. Considerations include a holistic approach to the child with a DSD, inclusion of the parents, recognition of psychological wellbeing, the role for support groups, and need for comprehensive longitudinal care. Outcome data collection, although still limited, expands medical knowledge and quality improvement. Development and evaluation of multidisciplinary care models focusing on quality of care is ongoing.
Talking with the parents
For parents, the birth of their child is a long-anticipated and exciting event. Because of the increased frequency of prenatal ultrasound examinations, parents have usually been told the sex of their child, may have selected their infant’s name, and often hosted a “gender reveal” celebration. In some instances, abnormal genital development has been identified through ultrasound and the parents have been informed of this situation. In this scenario, parents and healthcare providers can confer regarding the likely diagnostic testing and treatment recommendations. In the absence of prior knowledge, the parents are suddenly confronted with a newborn with a birth defect involving the external genitalia and uncertainty regarding their infant’s sex. Anxiety and apprehension regarding the baby’s health and sex are particularly traumatic when parents had no prior awareness of the abnormal genital development.
Initially, the parents need to be congratulated on the birth of their child. They need to hear that their child has a condition affecting sex development and that this condition will be carefully and completely addressed. Appropriate discussions include sharing that abnormalities of sex development involve the complex system that directs the development of reproductive system, including external genital development. Explaining that the sex of their child cannot be established simply by examining the external genitalia is essential. It is important to emphasize that the atypical development is not the parents’ fault and that they should not feel guilty or ashamed. Physician and healthcare professionals should not speculate or offer presumptive diagnoses. The parents can be reassured that all appropriate studies will be obtained to provide comprehensive information to best establish their child’s sex of rearing. Use of genetic tools may ascertain the specific etiology of the DSD, with the caveat that a specific molecular etiology may remain unknown.
The goal is patient-centered care within the context of the family and a multidisciplinary team that includes pediatric endocrinologists, pediatric urologists/surgeons, geneticists, neonatalogists, radiologists, behavioral health providers, and pediatric endocrine nurse educators. An initial treatment goal is to ascertain whether an underlying or associated life-threatening condition requires specific urgent treatment. One team member should serve as the principal communicator with the family.
Although contemporary society includes overt references to sex and sexuality, parents may have difficulty thinking of their child as a sexual being and feel embarrassed discussing their child’s sex, gender identity, and future sexuality. Cultural attitudes, preexisting expectations, and family support systems impact the parent’s responses and may influence their choices for the infant. Parents need to be informed that the child’s gender identity is a personal experience of one’s gender. The medical team needs to promote an open and caring network to provide support for the parents. Importantly, the medical team needs to engage the parents in the medical decision-making process and discuss pertinent information. It is important to be aware that the clinicians’ use of clinical uncertainty may misdirect parents and exacerbate their anxiety. For surgical decision-making, clinicians should strive for balance between caution and benefit and consideration of options beyond the dichotomy of “surgery” versus “no surgery.
If the child’s sex remains unclear, information needs to be obtained to assist the parents and healthcare team to determine the most appropriate sex of rearing. Usually, this can be accomplished within a matter of hours or days. In more complex instances, the diagnostic process may take longer. In situations in which it is impossible to identify the specific etiology, the general DSD category (see later) provides a basis for decision-making.
Factors relevant for the medical decision-making process include the extent of external and internal reproductive system development, evidence of gonadal functionality (potential for pubertal hormone secretion and fertility), and hormone responsiveness. In some instances, these factors are more relevant than the karyotype. Genes and gene products involved in sex development are mapped to autosomes and sex chromosomes. Genetic variants mapped to coding and noncoding regions influence sex differentiation and sex development of the fetus and child.
When consensus has been reached regarding a diagnostic category, available outcome information should be reviewed. Knowledge of a specific etiology, including immediate details and long-term outcome, enables optimal planning of therapeutic interventions and genetic counseling for future pregnancies. Healthcare providers need to be cognizant that available outcome data to assist in the decision-making processes are limited. Currently accessible information in published reports is largely based on retrospective studies obtained using diverse methods and strategies. The extent, timing, and duration of prenatal androgen exposure, which can only be estimated, likely influences development of the central nervous system (CNS) and other tissues. Prenatal androgen exposure may influence development of gender identity.
One multicenter cross-sectional study reported short-term outcome data on 92 children found to have genital ambiguity with karyotype distribution of 57%, 46,XX; 34% 46,XY; and 9% sex chromosome anomalies. Over 90% of 46,XX individuals had congenital adrenal hyperplasia (CAH); 65% of the 46,XY individuals lacked a molecular diagnosis; and most 46,XY individuals were being raised as males. Changing concepts regarding medical and surgical interventions urge caution regarding decision-making, while additional outcome studies are underway. Studies may address specific aspects, such as compliance with hormone therapy and patient satisfaction or psychological health, quality of life, and self-perceptions among males with CAH.
The first conversation with the parents should reinforce parental bonding with their infant. As much as possible, a positive and optimistic tone is helpful. Indeed, the emotional tone of this initial conversation is more meaningful than the factual information provided and is recalled by parents for many years. Respect for the family and individual viewpoints, together with a willingness to repeat or defer detailed explanations, are crucial. In the midst of the emotional distress associated with the uncertainty of their infant’s sex, parents may be unable to assimilate the vast amount of information that eventually needs to be shared. Repeated discussions with the parents will enable them to acknowledge their emotional and intellectual concerns regarding their infant. Familiarity and understanding will enable the parents to bond with their infant and interact with family members, friends, and colleagues.
Unless the natal sex is clear at this point, the healthcare team should recommend that the parents delay naming the infant, announcing the baby’s birth, and registering the birth, until more information becomes available. The message should be clear that they will be actively involved in the process to establish the sex of rearing for their child. Until sex of rearing is established, it is best to refer to the infant as “your baby” or “your child.” Terms such as he , she , and it should be avoided. The multidisciplinary team is responsible for educating ancillary staff regarding how to refer to the infant as “your baby.”
Factual explanations regarding the process of sexual differentiation with a focus on their infant’s situation should be initially outlined. The primary goal at this point is to provide the parents with a basic understanding that the internal and external genital structures for both boys and girls develop from the same primordial tissues. It is also helpful to explain that there are no exclusively male and female hormones. Rather, the environments in which male and female fetuses develop are characterized by differing relative amounts of these hormones. Using simple sketches, pictures, and diagrams can be helpful to explain the embryology of genital development to the parents. Some parents may benefit from practicing the words they will use to discuss the infant’s health with other family members. Detailed explanations can be reviewed multiple times as the child ages, especially because the child is unable to actively participate in the initial discussions.
During the early conversations, examining the infant with the parents to identify the specific physical findings of their infant is often beneficial. Viewing their child’s external genitalia can reduce their apprehension and reinforce the perception that their infant’s needs are similar to all infants. Information can be presented to minimize anxiety and better equip parents to participate in the decision-making process. Discussion of the many concerns (particularly those related to gender identity, pubertal development, sexual orientation, sexual function, and fertility) may be helpful. Honestly addressing parents’ concerns will ultimately create trust and positive feelings to help the parents promote their child’s self-esteem. Ideally, each parent achieves a personal resolution with a commitment to a positive perspective for the child’s future.
Terminology
Under the auspices of the Pediatric Endocrine Society (NA) and the European Society for Pediatric Endocrinology, an international consensus statement was formulated that recommended a revised classification of the medical terminology used for disorders of sex development to avoid confusing and derogatory terms. This descriptive classification attempts to be sensitive to concerns of parents and flexible enough to incorporate novel molecular genetic information. The updated classification system integrates molecular genetic considerations into the nomenclature for “disorders of sexual differentiation (DSD)” and provides an approach to the diagnostic evaluation. There are objections to use of the word “disorders” because this implies pathology, with “differences” sometimes being used.
Terms, such as pseudohermaphrodite, true hermaphrodite , and gender labeling in the diagnosis should be eliminated. However, some patients still prefer to use the term intersex . To accommodate all types of DSD, the classification system is broad and includes some conditions that do not present with obvious abnormalities of genital development ( Table 6.1 ). The primary goal of this classification system is to provide a framework for diagnosis, assessment, and care management based largely on sex chromosome status. Currently, microarray, candidate gene analyses, and whole exome/genome sequencing are increasingly used. The DSD categories include sex chromosome DSDs, such as 45,X/46,XY; ovotesticular DSD; 46,XY DSDs, such as disorders of testicular development, disorders of androgen synthesis and action, 46,XY sex reversal, and 46,XX DSDs; and 46,XX sex reversal. Some diagnoses are included in more than one category because of the complexities of chromosomal and gonadal development. The number of genes identified to be involved in sex development continues to increase. Nevertheless, despite many recent advances, the specific molecular etiology of the genital ambiguity in an individual cannot always be identified, especially among those with 46XY DSDs.
| Syndrome | Karyotype | Chromosomal Defects |
|---|---|---|
| Turner syndrome | 45,X mosaic 45,X/46,XX | X monosomy Mosaic monosomy X |
| Turner syndrome with structural X chromosome rearrangements | 46,X,i(Xq) 46,X,del(Xp) 46,X,+mar | isochromosome Xq deletion Xp marker chromosome |
| Gonadal dysgenesis | 45,X/46,XY XX/XY | Mosaic loss of the Y chromosome in XY, chimerism |
| XX male SRY gene positive Gonadal dysgenesis | 46,XX or 46,X,der(X)t(X;Y) | Yp ( SRY gene) translocation to the X chromosome or autosome |
| XX male SRY gene negative | 46,XX | SRY gene is absent |
| Klinefelter syndrome and its variants | XXY mosaic 46,XXY/46,XY XXYY XX/XXY | Disomy X, Mosaic disomy X Disomy X and Y Mosaic loss of the Y chromosome in XXY |
Sex determination
Through Alfred Jost’s experiments with fetal rabbits in the 1940s and 1950s, the critical requirements for a testis and testosterone for male sexual differentiation were established. Chromosomal composition of the human embryo, XX or XY, determines gonadal sex. Investigation of human sex chromosome evolution suggested that the human sex chromosomes evolved from a pair of ancestral autosomes approximately 300 million years ago. The emergence of the SRY locus, several Y chromosome inversions, and loss of X-Y crossing-over led to the current human situation.
Studies of patients with disorders of sex development ultimately led to identification of the genetic locus primarily responsible for this binary switch, the sex-determining region on the Y (SRY) gene on the Y chromosome. Studies involving creation of transgenic SRY + mice confirmed the essential role of SRY and provided further molecular understanding of testicular differentiation.
Genetic sex is established at fertilization followed by sex determination, the binary switch that launches the developmental destiny of the embryonic gonads to become testes or ovaries. Sex determination is largely influenced through transcriptional regulation, whereas secreted hormones and hormone receptors influence phenotypic development. Sexual differentiation refers to the process through which male or female phenotype develops.
The gonads, internal genital ducts, and external genital structures all develop from bipotential tissues. Each cell in the developing gonad has the potential to differentiate into either a testicular or ovarian cell, depending on how the transcriptome of the undifferentiated cell realizes its pathway to develop into an ovary or testis. In this bipotential state, pluripotency with genes poised for either activation or repression exists. In other words, sex determination of the bipotential gonads depends on cell fate commitment to one pathway at a precise moment in development, while maintaining active repression of the alternative developmental pathway.
In the usual situation, the karyotype (46,XY or 46,XX) of the primordial gonad determines whether it differentiates into a testis or ovary, respectively. Inherent differences exist between XX and XY cells; approximately 85% of the second X chromosome in an XX cell undergoes X-inactivation and Y chromosome genes related to spermatogenesis are expressed in XY cells. Local factors (such as hormones secreted by the developing gonads or tissue-specific transcription factors) influence the ensuing differentiation of the internal and external genital structures. This process integrates sex-specific pathway signals that appear to antagonize each other.
Chromatin configuration and its spatial three-dimensional architecture influence gene expression by modulating the ability of transcription factors to bind to deoxyribonucleic acid (DNA). Histone modification and DNA methylation influence chromatin organization. The chromatin landscape actively modulates the unending ovary versus testis antagonism. Genes involved with male pathway development lose their repressive marks when the testis pathway is activated and vice versa for genes involved in the ovarian pathway. Cis-regulatory elements, such as silencers and enhancers, coordinate the specific spatiotemporal expression of genes within the transcriptional network; sex-specific regulatory elements are acquired during development. Polycomb proteins are conserved transcriptional regulators that coordinate chromatin structure and chromosome architecture. Chromatin regulators, for example, polycomb proteins, represent critical nodes where biological signals modulate gene expression.
Divergence from the normal sequence of events leads to disorders of sex development that can manifest as abnormal gonadal differentiation, inconsistent internal genital differentiation, or ambiguity of the external genitalia. Although genital ambiguity is usually not considered to be a medical emergency, this type of birth defect is usually extremely distressing to the parents and extended family. When adrenal insufficiency accompanies the genital ambiguity, immediate evaluation and treatment are essential. Regardless, prompt referral and evaluation by a multidisciplinary team with expertise in disorders of sex differentiation is strongly recommended.
Development of the reproductive system
Urogenital Ridge and Bipotential Gonad Development
The urogenital ridge gives rise to the gonads, adrenal cortex, kidney, and reproductive tract. The gonads are derived from intermediate mesoderm and depend on the correct ingression, proliferation, and orientation of the coelomic epithelial cells ( Fig. 6.1 ). In humans, at 4 to 6 weeks of gestation, the urogenital ridges develop as paired outgrowths of coelomic epithelium (mesothelium) on the ventral side of the mesonephros. As the coelomic epithelium proliferates, its basement membrane disintegrates to allow ingression of coelomic cells to form the developing gonad. Notch signaling ensures proper polarization of these cells. In the developing testis, the ingressing cells initially form the Sertoli cells, whereas the later ingressing cells develop into the interstitial cells, including Leydig cells. In the ovary, ingressing cells give rise to both theca and granulosa cells.
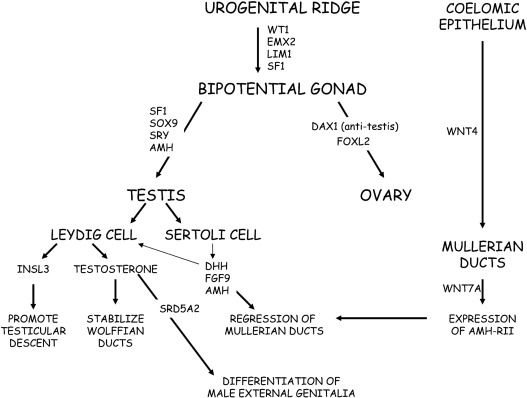
The somatic cells of the bipotential gonad require expression of Wilms tumor ( WT1) , GATA binding protein 4 (GATA4), and steroidogeneic factor 1 (NR5A1/SF1 ) genes. The WT1 gene codes a zinc transcription factor, which is expressed in embryonic mesodermal tissues and appears to influence mesodermal-epithelial interactions. GATA4 is expressed in the somatic cells of the urogenital ridge and bipotential gonad before showing sex-specific expression. Chromobox homolog 2 ( CBX2 ) is expressed in the developing gonad at 7 weeks of gestation and plays a role in early gonadal development. CBX2 functions upstream of SRY , promotes transactivation of NR5A1 and SOX9, and represses forkhead transcription factor 2 (FOXL2) . NR5A1 is expressed in the urogenital ridge and appears to upregulate SRY expression, furthering testis development. FOXL2, a forkhead transcription factor expressed in the ovary, orchestrates cell fate decisions; FOXL2 favors ovarian development and represses testicular development. In addition to transcription factors and specific secreted factors (hormones), physical contact with the mesonephros appears to be important for subsequent gonadal differentiation.
Because of their origin within the developing urogenital system, ovaries, and testes are initially located high in the abdomen near the kidneys. Specific signaling molecules activate or repress gonad determination for both testes and ovaries. As noted earlier, ongoing mutual antagonism or competition between specific genes and proteins influences cell fate decisions in gonad development. Transcription factors modulate this “competition” by influencing gene expression. Specific examples include FOXL2 versus SRY-box 9 ( SOX9 ) and SOX9 versus Wingless-type MMMTV integration site family member 4 ( WNT4 )/β-catenin, which are discussed later. Hence, sex determination and differentiation reflect mutual antagonism between the testes determining factors , SRY-SOX9-FGF9 , and the ovary determining factors, roof plate-specific spondin-1 ( RSPO1)-WNT -β-catenin- FOXL2 . Curiously, the antagonism persists postnatally (see later). As would be anticipated, steroidogenic enzyme genes show higher expression in developing human testes compared with ovaries.
Human Testicular Development
Testicular differentiation begins earlier than ovarian development. The first evidence of testicular differentiation is the appearance of primitive Sertoli cells at 6 to 7 weeks’ gestation in the human fetal testis. Cells, mostly endothelial cells, migrate from the mesonephros and interact with the pre-Sertoli cells to promote development of the testicular cords, which is initiated at approximately 7 to 8 weeks of gestation with identifiable cords by 9 to 10 weeks of gestation. The testicular cords are precursors of the seminiferous tubules that will contain Sertoli and germ cells. Interactions between endothelial and mesenchymal cells appear to influence development of the testicular cords.
The binary switch responsible for testicular development is the SRY gene located on the short arm of the Y chromosome. The precise mechanism(s) responsible for triggering SRY expression are incompletely defined. In the human testis, SRY is expressed in supporting cells at approximately 6 weeks of gestation; low-level expression persists throughout gestation. The SRY protein contains a high-mobility group (HMG) domain and is encoded by a single exon gene. Two nuclear localization signals are located in the HMG domain. The SRY protein is expressed in pre-Sertoli cells, where it triggers a molecular switch to induce Sertoli cell differentiation, thus, initiating the process of male sexual differentiation. The HMG domain of the SRY protein binds to the minor DNA groove, where it functions as a transcription factor by bending DNA to presumably permit other proteins access to regulatory regions and to promote assembly of nucleoprotein transcription complexes. A threshold SRY level must be achieved at a critical time during gestation to establish male sexual differentiation. Otherwise, the ovarian differentiation pathway is activated.
SRY expression is independent of the presence of germ cells. SRY increases the expression of the SRY-related HMG box-containing-9 ( SOX9 ) gene. Phenotype-genotype studies of humans and mice demonstrate that SOX9 expression is a crucial step, downstream of SRY , in testis development. Upstream from the SOX9 transcription start site, at least three enhancers, eSR-A, eSR-B, and eALDI, synergize to promote SOX9 expression. Available data suggest that SRY and NR5A1 bind at eALDI to promote initial SOX9 expression. Once expression has been initiated, SOX9 positively autoregulates its own expression. Subsequently, SOX9 and NR5A1 bind to regions within all three enhancer loci to amplify testicular SOX9 expression. Using both direct and indirect mechanisms, SOX9 interferes with genes promoting ovarian differentiation. These data are consistent with the suggestions that mammalian gene regulation depends on multiple redundant enhancers and that copy number variants in this region may cause sex reversal.
In addition to SRY , SOX9 , and NR5A1 , expression of other genes is required for normal male sexual differentiation. Fibroblast growth factor (FGF9) acting through FGF receptors (FGFRs) promotes expression of SOX9 and repression of WNT4 and FOXL2 . Genes found to have expression pattern similar to SOX9 during testis development include CITED1, ANKRD18A, G6PD, SLC52A3, KEL, ZNF280B, PRPS2, and INHBB . In addition to activation of the testicular differentiation pathway, the developing testis expresses factors to actively repress ovarian development. Specifically, the developing testis represses the R-spondin-WNT signaling pathway. Transmember E3 ubiquitin ligases, ZNRF3 and RNF43, inhibit WNT signaling by targeting the Frizzled receptor for degradation.
By immunohistochemistry, NR5A1 and SOX9 proteins can be detected in human embryonic gonadal tissue at 6 to 7 weeks of gestation. At this time, SOX9 expression becomes limited to nuclei of Sertoli cells in a 46,XY fetus but remains cytosolic in a 46,XX fetus. Subsequently anti-Mullerian hormone (AMH) is expressed; with increased AMH protein expression Wilms tumor (WT1) and GATA-4 protein expression increase in the fetal testis. Male-limited expression of doublesex-related and Mab-3–related transcription factor 1 (DMRT1 ) was found in 6- and 7-week-old human fetuses. DMRT1 appears to be necessary to maintain testicular cords and prevent transdifferentiation to a female phenotype. The most rapid growth in Sertoli cell number appears to occur during the latter half of the first trimester and the second trimester. After Sertoli cells have developed, fetal Leydig cells appear around the seventh to eighth week of gestation and produce androgens to promote internal and external male genital structures.
Leydig cells are comprised of two distinct populations; fetal Leydig cells differentiate in utero and adult Leydig cells emerge after birth. Fetal Leydig cell differentiation depends on paracrine signals, such as platelet-derived growth factor receptor-alpha (PDGFR-α), Desert hedgehog (DHH), PTCH1, and Aristaless-related homeobox (ARX). NR5A1 is expressed in Leydig cells to promote steroidogenic enzyme gene expression. The number of fetal Leydig cells reflects gonadotropin stimulation because the number is decreased in anencephalic male fetuses and increased in 46,XY fetuses, with elevated gonadotropin concentrations secondary to complete androgen insensitivity.
Expression of genes associated with testicular steroidogenesis increases around 8 weeks of gestation. Placental human chorionic gonadotropin (hCG) stimulates early testicular androgen secretion. Later in gestation, pituitary LH secretion drives testicular androgen secretion. By 11 weeks of gestation, the testicular compartments, tubular and interstitial components, and the cell types of interest (Leydig, Sertoli, and germ cells) can be visualized. In human fetal testes, HSD17B3, CYP11A1 , and PTC1 messenger ribonucleic acid (mRNA) levels increased significantly through the second trimester without significant changes in CYP17A1, LHR, or INSL3 levels.
Human Ovary Development
Rather than ovarian differentiation being the default pathway occurring in the absence of SRY gene expression, it is clear that specific genes influence ovarian differentiation. Genes involved in ovarian differentiation include WNT4 , FOXL2 , follistatin ( FST ), bone morphogenic protein-2 ( BMP2 ), GATA4/FOG2 , and RSPO1 . RSPO1 is a secreted factor that activates the β-catenin WNT signaling pathway. FOXL2 represses male-specific genes, especially SOX9 beginning in the fetus; this active repression continues throughout adulthood. Hence FOXL2 and SOX9 expression are mutually exclusive in developing gonads, as well as postnatal ovaries and testes.
Normal ovarian development requires FOXL2, RSPO1, and canonical WNT/β-catenin signals. RSPO1 acting through LGR4/5 cell-surface receptors sequesters the transmembrane E3 ubiquitin ligases ZNRF3 and RNF43, resulting in increased WNT signaling. RSPO1 and WNT4 stabilize and amplify β-catenin signaling to activate target gene transcription. WNT4 suppresses androgen-secreting interstitial cells, inhibits coelomic vascularization, and supports Mullerian derivatives.
Two genes that factor in germline development, alpha ( FIGLA ) and newborn ovary homeobox ( NOBOX ), are expressed in fetal or neonatal germ cells, where they recruit developing granulosa cells to form primordial and primary follicles. Additional transcription factors involved in ovarian development include SOHLH1, SOHLH2, NOBOX, LHX8, FIGLA, and LHX9. The second trimester human fetal ovary expresses proteins necessary to synthesize and respond to estrogenic, progestogenic, and androgenic signaling.
Germ Cell Development
Despite the prerequisite for germ cell development to perpetuate the species, germ cells are not required for the initial development of ovaries or testes. The primordial germ cells originate in the epiblast. At approximately 6 weeks of gestation in the human, the primordial germ cells proliferate and migrate from the hindgut along nerve fibers to colonize the genital ridges. SOX17 is a pluripotency factor that is critical for human primordial germ cell specification through its action to promote germ cell–specific gene expression. SOX17 appears to promote BLIMP expression, which appears to suppress endodermal and mesodermal genes.
Upon reaching the developing gonad, the local environment directs the fate of the primordial germ cells toward male or female development. Hence the terminal differentiation pathways for germ cells are sexually dimorphic. When this migration process goes awry, the gonadal germ cell population may be deficient. Aberrant migration can result in ectopic migration into other organs. Ectopic germ cells are primarily located in the CNS, but can also be found in the mediastinum, thorax, and pelvic region. Germ cell tumors may develop in ectopic germ cells when the germ cells are not eliminated by apoptosis.
Developmental phases for female germ cells include initiation of meiosis, formation and breakdown of germ cell nests, and assembly of single oocytes into primordial follicles. For male germ cells, the characteristic phases are migration, mitosis, and cell-cycle arrest. Oocytes undergo meiosis, whereas meiosis is actively inhibited in male germ cells. The RNA binding protein deleted in azoospermia-like (DAZL) appears to license entry into meiosis; DAZL expression increased between 9 and 14 weeks’ gestation. Several RNA-binding proteins, such as LIN28, DAZL, and BOLL, are expressed during oogenesis. Another protein, meiosis specific with coiled-coil domain (MEIOC) stabilizes mRNAs that encode proteins relevant for meiosis; its expression appears to be independent of retinoic acid. DAZL inhibits SOX17 expression limiting germ cell pluripotency. Hence DAZL plays dual roles: it initiates meiosis in ovaries and represses pluripotency factors.
The fetal ovary is characterized by a developmental gradient with existence of multiple subpopulations of germ cells at different developmental stages; the more differentiated germ cells are located in the center of the developing ovary. In the developing ovary, the germ cells initially form oogonial clusters connected by intracellular bridges. Selected oogonia enter meiosis and progress through meiotic prophase I (MPI). Somatic cells surround individual oocytes to form follicles. Around 16 to 20 weeks’ gestation, the oogonial clusters break down to form primordial follicles. The oocytes within the primordial follicles undergo growth arrest at the diplotene stage, lasting until oocyte growth is reinitiated with the onset of puberty. The greatest number of follicles exists at midgestation. From a peak of 6.8 million oocytes at approximately 5 months of gestation, approximately 2 million are present at birth. Notch signaling mediates interactions between oocytes and granulosa cells, regulates oocyte survival, and promotes breakdown of the oogonial nests. The internal environment throughout fetal ovary development has the potential to directly influence the fertility of the developing female fetus (controlling the size of the ovarian reserve) and the quality of the oocytes that will eventually become her child (by influencing the extent of selection and apoptosis).
The primordial follicles constitute the ovarian reserve. Postnatally, recruitment and maturation of the primordial follicles is largely governed by AMH, androgen, luteinizing hormone (LH), and follicle-stimulating hormone (FSH) secretion. Premature germ cell loss can lead to gonadal dysgenesis or premature ovarian insufficiency (POI). Accelerated follicular atresia contributes to the follicular depletion characteristic of streak gonads in X monosomy. Although not typically associated with genital ambiguity, mutations in genes governing oocyte and ovarian development cause DSDs characterized by delayed puberty or POI. These genes influence germ cell mitosis, germ cell meiosis, and DNA repair.
In contrast, testicular germ cell development is less dynamic. The Sertoli cells envelope the germ cells to form seminiferous cords at approximately 7 to 9 weeks of gestation in the human XY gonad. The germ cells in the developing testis enter a state of mitotic arrest until the onset of puberty.
Early during gestation, primordial germ cells undergo epigenetic reprogramming, which modifies chromatin architecture. In other words, inherited imprints are erased and DNA methylation is decreased at CpG islands, transcription start sites, genes, and intergenic regions. Demethylation affects almost the entire genome, including the inactive X chromosome by 10 to 11 weeks of gestation. During meiosis, both X chromosomes need to be active to enable efficient pairing. Subsequently, maternal and paternal alleles undergo sexually dimorphic imprinting, such that developing oocytes and sperm are differentially marked reflecting “parent-of-origin”-specific methylation patterns. This process occurs late in fetal development in the male and postnatally in female germ cells. Study of parent-of-origin-dependent disorders, such as Beckwith-Wiedemann, Prader-Willi, and Angelman syndromes and neonatal diabetes mellitus has elucidated the importance of this imprinting process.
Human Adrenal Development
By 33 days postconception, the human fetal adrenal cortex is distinct from the developing gonad. Because of its role as the source of dehydroepiandrosterone sulfate (DHEA-S) for placental estrogen biosynthesis, the fetal adrenal cortex grows rapidly. By 50 to 52 days postconception, expression of several steroidogenic enzymes, steroidogenic acute regulatory protein ( StAR ), 11β-hydroxylase ( CYP11B1 ), 17α-hydroxylase/ 17,20-lyase ( CYP17A1 ), and 21-hydroxylase ( CYP21A2 ) in the fetal adrenal cortex have been demonstrated immunohistochemically. Transitory fetal adrenal cortisol biosynthesis peaks at 8 to 9 weeks’ gestation, which coincides with transient adrenal expression of both nerve growth factor IB-like ( NGFI-B ) and 3β-hydroxysteroid dehydrogenase-2 ( HSD3B2 ). At the same time, adrenocorticotropin hormone (ACTH) can be detected in the anterior pituitary, suggesting the presence of negative feedback inhibition during the first trimester. During the time male sexual differentiation begins, this negative feedback inhibition may serve to prevent virilization of female fetuses to ensure normal female sexual differentiation. Distinct expression profiles for fetal adrenal steroidogenic enzymes have been described with differences between the first and second trimesters. Chromaffin cells derived from the sympathetic nervous system migrate into the adrenal cortex to form the adrenal medulla.
Development of Internal Genital Structures
The Wolffian duct originates as the excretory duct of the mesonephros and develops into the epididymis, vas deferens, ejaculatory duct, and seminal vesicle. The Mullerian or paramesonephric duct originates as an invagination of the coelomic epithelium and develops into the Fallopian tubes, uterus, and upper third of the vagina.
The Sertoli cells of the developing testis secrete AMH, also known as Mullerian inhibitory hormone ( MIH ). In human 46,XY fetuses, AMH expression can be detected by 7 weeks of gestation, is not dependent on the presence of germ cells within the testis, and promotes regression of the Mullerian ducts. AMH, a member of the transforming growth factor-β (TGF-β) family, undergoes proteolytic cleavage to become biologically active. AMH binds to its receptor, AMH-RII, on the surface of the Mullerian duct mesenchymal cells to induce increased matrix metalloproteinase 2 expression. The net result is degeneration and loss of basement membrane integrity of the epithelial and mesenchymal Mullerian cells, leading to regression of the Mullerian ducts.
AMH expression is highly regulated; inappropriate expression in a 46,XX fetus would lead to uterine agenesis. In the 46,XX fetus with absence of both AMH and testosterone, the Mullerian duct derivatives persist and the Wolffian ducts regress. When a female fetus is inappropriately exposed to AMH (as in freemartin cattle), Mullerian duct regression and ovarian masculinization occur. The paired Mullerian ducts arise as coelomic epithelial invaginations. Around the eighth week of gestation, the Mullerian ducts fuse, followed by degeneration of a midline epithelial septum. Variable persistence of this septum can lead to uterine malformations. Multiple factors influence the development of the uterus, cervix, and vagina. HOX genes specify the development program for distinct uterine regions; HOXA9 specifies the uterine tube, HOXA10 and HOXA11 specify the uterus, and HOXA13 specifies the vagina. The vaginal plate appears during the 11th week of gestation through occlusion of the Mullerian-derived uterovaginal canal and ultimately disappears as the vaginal plate lengthens and canalizes.
The fetal hypothalamic-pituitary-gonadal (HPG) axis is active by midgestation, with peak fetal testosterone concentrations occurring at approximately 15 to 16 weeks of gestation. Before this time, placental hCG stimulates testosterone production by the fetal Leydig cells. Secretion of testosterone by the fetal Leydig cells stabilizes the Wolffian ducts in 46,XY fetuses. Region-specific signaling molecules, such as BMPs, homeobox (HOXA10 and HOXA11), growth differentiation factor 7 (GDF7), relaxin, an orphan G-protein–coupled receptor (LGR4), platelet-derived growth factor A (PDGFA), and its cognate receptor (PDGFRA) influence the development of the epididymis and seminal vesicle.
The prostate, a male accessory sex gland, contributes to seminal fluid plasma and develops from the urogenital sinus. After the initial testosterone-dependent induction of prostate differentiation, subsequent development involves epithelial-mesenchymal interactions that lead to cell differentiation and branching morphogenesis. The requisite signaling molecules, FGFs, sonic hedgehog (SHH), BMPs, HOXA13, and HOXD13, are similar to those required for external genital differentiation.
Development of External Genital Structures
The genital tubercle, urethral folds, and labioscrotal swellings give rise to the external genitalia. Androgens play a time-dependent role in formation, differentiation, and growth of the fetal male external genitalia at approximately 8 to 14 weeks of gestation, during the masculinization programming window (MPW).
During this period, a cylindrical 2-mm phallus with genital swellings develops by 9 weeks of gestation. Within the developing penis, the urethra forms by canalization of the urethral plate with subsequent fusion to form the penile urethra by 12 to 14 weeks of gestation. The genital tubercle develops into the corpora cavernosa of the penis, and the labioscrotal folds fuse to form the scrotum. By 14 weeks, the external genitalia are clearly masculine, apart from testicular location. Thereafter, the penis grows at a rate of 0.7 mm/week from 14 weeks to term. Deficiency of androgen or androgen action during the MPW reduces penis length, an effect that cannot be fully rescued by postnatal T therapy. The MPW predetermines potential phallic size, whereas postnatal androgen action is required to realize normal potential.
In the 46,XX fetus, in the absence of androgens, the urethral folds and labioscrotal swellings do not fuse and develop into the labia minora and labia majora, respectively. The genital tubercle forms the clitoris, and canalization of the vaginal plate creates the lower portion of the vagina. By 11 weeks of gestation, the clitoris is prominent and the lateral boundaries of the urogenital sulcus have separated. Minimal clitoral growth, well-defined labia majora, hypoplastic labia minora, and separate vaginal and urethral perineal openings are present by 20 weeks of gestation.
Anogenital Distance
The anogenital distance (AGD), the distance from anus to the genital tubercle, indicates prenatal androgen action and, as anticipated, is sexually dimorphic. Investigators have used different anatomic landmarks to assess AGD in humans. Salazar-Martinez and colleagues defined AGD as being from the anus to the perineoscrotal junction (anoscrotal distance) in males and from the anus to the anterior fourchette (anofourchettal distance) in females; this definition appears to improve interobserver reliability. Salazar-Martinez and colleagues reported normative ranges for anoscrotal and anofourchettal distances to be 21 ± 3.0 mm in males and 11 ± 2 mm in females (Salazar). In both sexes, AGD increased up to 12 months and maintained the sex dimorphic pattern. AGD also showed positive correlation with penile length at birth and with the increase in AGD from birth to 3 months. AGD provides a biomarker of early fetal androgen exposure and masculinization. Measurement of AGD can be used as a diagnostic tool for in utero androgen exposure and assessing impact of potential environmental endocrine disruptors on external genital development. AGD at birth predicts adult anogenital distance, but correlations with adult male reproductive parameters are inconsistent.
Sexual Differentiation of the Brain
Clinical investigations suggest that the brain is sexually dimorphic and that testosterone is a masculinizing hormone in human. Males with aromatase deficiency manifest male psychosexual behavior and gender identity. Alternatively, 46,XY individuals with complete androgen insensitivity syndrome (CAIS) develop female gender identity. However, preliminary data implicate genetic differences, independent of sex steroid exposure, as the molecular basis for some aspects of sexual dimorphism of the brain and other nongonadal tissues. Androgens have organizational and activation effects on CNS function. Complex interactions between genetic factors and hormone contribute to human brain sex differences.
Mouse models
For many patients with DSDs, the specific molecular etiology is unknown. Hence investigation of animal models, especially transgenic mouse models, has been helpful to delve into the processes involved in sex development. Investigation of normal and transgenic mice confirmed the crucial role of the sex-determining region on Y ( SRY ) gene in male differentiation when XX mice carrying only a 14-kb fragment of the Y chromosome showed a male phenotype. However, the molecular mechanisms responsible for sex differentiation/sex development and postimplantation development differ among rodents and humans. Despite these limitations, information gleaned from preclinical models provides useful information regarding molecular mechanisms of sex development.
Understanding gene regulation, epigenetic influences, and protein-protein interactions are essential to clarify regulation of the sex development pathways; this knowledge may identify factors relevant for patients with DSDs. The outcome of cell fate decisions involves antagonism between the male and female developmental programs. During the bipotential phase, both male- and female-specific genes are expressed. Subsequently, precise spatiotemporal expression of specific genes ensues. Abnormal gonadal development has been described in mice homozygous for targeted deletions of genes involved in urogenital differentiation, that is, Wt1 , Sf1 , Emx2, Cbx2/M33 , Six1/Six4 , and Lim1/Lhx9 . The phenotype of Wt1 knockout mice includes embryonic lethality, failure of gonadal and kidney development, and abnormal development of the mesothelium, heart, and lungs. Homozygous deletion of Emx2 , a homeodomain transcription factor, results in an embryonic lethal phenotype associated with absence of kidneys, ureters, gonads, and genital tracts. As Wt1 expression is initially normal in the metanephric blastema of Emx2 knockout mice, Emx2 is likely downstream of Wt1 . Interestingly, adrenal gland and bladder development are normal in Emx2 knockout mice. Although the phenotype of specific knockout mice have been reported, details regarding the regulatory mechanisms governing gene expression are often lacking.
Epigenetic mechanisms modify chromatin architecture to reversibly regulate gene expression in a developmental maner. These mechanisms include histone modifications, that is, methylation/demethylation, acetylation/deacetylation, ubiquitination, and DNA methylation. The polycomb group proteins, consisting of multiple protein subunits, function to modify chromatin structure. One polycomb protein is Cbx2/M33. The phenotype of Cbx2 knockout mice includes male-to-female sex reversal and hypoplastic gonads for both sexes. Although the testes were small, overexpression of Sry or Sox9 in Cbx2 knockout mice rescued sex reversal in XY mice. Based on available animal studies, Cbx2 modulates expression of Sf1/Nr5a1 , Sry , Sox9 , Dax1 , Gata4 , Arx, and Dmrt1 in both sexes, and influences gonad proliferation and size.
Although the temporal expression patterns for human SRY and mouse Sry differ, understanding the regulatory mechanisms responsible for mouse Sry expression has been germane to human SRY . Epigenetic factors influence expression of the mouse Sry gene. Both demethylation and histone modification are essential for Sry expression. The histone demethylase JMJD1A positively influences Sry expression by regulating H3K9me2 marks; Jmjd1a -deficient mice show partial male to female sex reversal. Another mechanism involves the GADD4G protein, which mediates DNA demethylation and mitogen-activated protein kinase (MAPK) signaling. Loss-of-function Gadd45g mutant mice manifest male-to-female sex reversal; GADD45G appears to activate the MAPK pathway by phosphorylating GATA4 to activate Sry expression. Mice with loss-of-function Gata4 mutation show phenotype similar to Gadd4g mice with decreased Sry expression and XY sex reversal.
Another epigenetic mechanism involved in Sry regulation involves CREB-binding protein (CBP, CREBBP, KAT3A) and p300 (EP300, KAT3B). These proteins are histone/lysine acetyl-transferases that modify chromatin-associated proteins to regulate gene expression. Mice with loss-of-function mutations manifest male-to-female sex reversal. These proteins can act as network hubs interacting with other proteins to influence gene transcription.
Investigation of mouse models has been used to learn more about Wolffian and Mullerian duct development. Development of Wolffian duct epithelium depends on signals from Wolffian duct mesenchyme. Testicular testosterone activates mesenchymal androgen receptors to initiate mesenchymal-epithelial cross talk, increase epidermal growth factor production, and antagonize COUP-TFII to promote Wolffian duct stabilization. Factors involved in Mullerian duct regression include AMH, AMHR2, and SMAD; however, the molecular mechanism(s) used by AMH to induce Mullerian duct regression remains to be defined.
Sex chromosome disorders
45,X/46,XY Mosaicism
Most sex chromosome disorders, such as 45,X and cytogenetic variants and 47,XXY and cytogenetic variants, are not associated with abnormal external genital development and will not be discussed in this chapter (see Table 6.1 ). In contrast, individuals with 45,X/46,XY and 46,XX/46,XY karyotypes manifest a broad range of phenotypes. Among individuals with 45,X/46,XY karyotype, internal and external genital structures range from normal male to ambiguous to female. Whereas the typical histological features consist of poorly developed seminiferous tubules surrounded by wavy ovarian stroma, gonadal differentiation can range from normal testis to streak gonads. At the time of puberty, virilization can occur.
The majority of individuals identified by prenatal karyotype as being 45,X/46,XY appear to be normally androgenized males; however, individuals diagnosed postnatally tend to have more clinical signs. A normal peripheral blood lymphocyte karyotype in individuals with gonadal dysgenesis suggests the presence of sex chromosome mosaicism within the gonad(s). Individuals with sex chromosome DSDs because of gonadal dysgenesis have an increased risk of developing gonadal tumors, such as gonadoblastoma or dysgerminoma because a dysgenetic gonad carrying a Y chromosome has an increased risk for neoplastic changes. Although gonadal tumors typically do not develop until the second decade of life, they can occur at younger ages.
Among a series of 63 males with 45,X/46,XY, subjects were classified into two groups; one group presented with genital anomalies and the second group presented with other reasons, such as short stature or infertility. Individuals identified by genital anomalies tended to have lower rates of spontaneous pubertal development, shorter stature, and greater likelihood of germ cell neoplasia. Although most gonads were classified as dysgenetic testes, some testes appeared relatively normal with evidence of spermatogenesis. No follicles were observed, suggesting that gonads labeled as ovotestis were mislabeled and consisted of undifferentiated/streak-like tissue. Based on these histological findings, it has been suggested that the etiology of the 45,X/46,XY karyotype is the loss of a Y chromosome in some cells. Focal spermatogenesis was identified in approximately 25% of these subjects. Thus when gonadectomy is considered because of risk for neoplasia, fertility preservation should be considered.
Disorders associated with disorders of sex development and additional congenital anomalies ( Table 6.2 )
ARX (X-Linked Lissencephaly 2)
X-linked lissencephaly 2 is characterized by genital ambiguity, lissencephaly, early-onset seizures, absence of the corpus callosum, intellectual disability, and hypothalamic dysfunction, and Aristaless-related homeobox ( ARX ) gene mutations. This gene is a member of the paired-type homeodomain transcription factor family and is mapped to Xp21.3. ARX contributes to many aspects of brain development including patterning, neuronal proliferation and migration, and cell maturation and differentiation, especially the generation and migration of GABAergic neurons. Findings in Arx knockout mice showed no expression in developing ovaries, but Leydig cell differentiation was impaired. Available data suggest that Arx is a positive regulator for the differentiation of fetal Leydig cells at the progenitor stage.
| Gene (CHR Band) | Syndrome | Karyo-Type | Genetic Defects | Mode of Inheritance | Characteristic Congenital Anomalies |
|---|---|---|---|---|---|
| ARX (Xp21.3) | Hydranencephaly with ABNORMAL GENITALIA (OMIM #300215) | XY | point mutations deletion/duplication | X-linked | lissencephaly, absent corpus callosum, early-onset intractable seizures, temperature instability |
| ATRX (Xq13.1) | Alpha-thalassemia mental retardation syndrome (OMIM # 301040) | XY | point mutations | X-linked | alpha-thalassemia, severe mental retardation, and genital abnormalities |
| CDKN1C (11p15) | IMAGe syndrome (OMIM # 614732) | XY | point mutations | AD maternal transmission | IUGR, metaphyseal dysplasia, adrenal insufficiency |
| CHD7 (8q12.2) | CHARGE syndrome (OMIM#214800) | XY XX | point mutations (70%) deletion/duplication (rare) | AD | coloboma, heart defects, choanal atresia, ear anomalies |
| DHCR7 (11q13.4) | Smith-Lemli-Opitz syndrome (OMIM #270400) | XY | point mutations (96%) deletion/duplication (4%) | AR | characteristic facial features, postaxial polydactyly, 2-3 toes syndactyly, cleft palate |
| EMX2 (10q26.11) | (OMIM # 600035) | XY | microdeletion | sex reversal, kidney, developmental delay,schizencephaly | |
| GLI3 (7p14.1) | Pallister-Hall syndrome (OMIM #146510) | XY XX | point mutations (20%–25%) deletion/duplication (rare) | AD | hypothalamic hamartoma, mesoaxial polydactyly, bifid epiglottis |
| HHAT (1q32.2) | chondrodysplasia (PMID: 24784881 ) | XY | point mutations | AR | dwarfism with generalized chondrodysplasia |
| HOXA13 (7p15.2) | Hand-foot-genital syndrome (OMIM#140000) | XY XX | polyalanine expansions (60%) point mutations (35%) deletion/duplication (2%–5%) | AD | shortening of the thumbs and great toes, bicornuate uterus |
| HSD17B4 a (5q23.1) | Perrault syndrome 1 (OMIM #233400) | XX | point mutations | AR | sensorineural deafness |
| KAT6B (10q22.2) | Genitopatellar syndrome (OMIM #606170) | XY | point mutations deletion/duplication (rare) | AD | patellar hypoplasia, club feet, microcephaly, flexion contractures, agenesis of the corpus callosum |
| POR (7q11.2) | Antley-Bixler syndrome (OMIM#201750) | XY XX | point mutations deletion/duplication (rare) | AR | radiohumeral synostosis, midface hypoplasia, choanal stenosis, joint contractures |
| RSPO1 (1p34.3) | Palmoplantar keratosis/sex reversal (OMIM #610644) | XX | point mutations | AD | palmoplantar keratoderma, predisposition to squamous cell carcinoma of the skin |
| SAMD9 ( 7q21.2) | MIRAGE syndrome (OMIM # 610456) | XY | point mutations | AD | myelodysplastic syndrome, IUGR, adrenal insufficiency, chronic diarrhea, pulmonary infections |
| SOX9 (17q24.3) | Campomelic dysplasia (OMIM #114290) | XY | point mutations (90%) translocation (5%) deletion/duplication (2%) | AD | Pierre Robin sequence, cleft palate, short bowed limbs, 11 pairs of ribs |
| TBX3 (12q24.1) | Ulnar-mammary syndrome (OMIM #181450) | XX | point mutations | AD | ulnar limb deficiencies, mammary gland hypoplasia, dental abnormalities |
| TP63 (3q28) | Limb-mammary syndrome (OMIM #603543) | XX | point mutations | AD | hand/foot anomalies, mammary gland and nipple |
| TSPYL1 (6q22.1) | SIDDT (OMIM #608800) | XY | point mutations | AR | bradycardia, hypothermia, laryngospasm, bronchospasm |
| WT1 (11p13) | Denys-Drash, Frasier, and Meacham syndromes (OMIM #194080) | XY XX | point mutations | AD | Wilms tumor, nephropathy, gonadoblastoma, congenital diaphragmatic hernia |
| ZEB2 (2q22.3) | Mowat-Wilson syndrome (OMIM#235730) | XY | point mutations (80%–85%) deletion/duplications (15%–17%) translocations (1%–2%) | AD | Hirschsprung disease, congenital heart defects, agenesis of the corpus callosum, microphthalmia, Axenfeld anomaly |
CDKN1C (IMAGe Syndrome)
The IMAGe syndrome is characterized by intrauterine growth retardation, metaphyseal dysplasia, adrenal hypoplasia, cryptorchidism, and micropenis. Genital anomalies appear to be limited to affected males. Both sporadic and familial cases have been described. Heterozygous missense mutations in the CDKN1C gene have been detected in some cases. This gene is located at chromosome 11p15.5, encodes a 316-amino-acid protein, p57 Kip2 , negatively regulates cellular proliferation, and inhibits cell cycle progression. The gene is located within a complex genetic locus containing several genes including, IGF2 and H19 , subject to imprinting modulation through cis-acting elements; CDKN1C is maternally expressed. Mutations associated with the IMAGe or Silver-Russell syndromes appear to be localized to the proliferating cell nuclear antigen–binding domain, resulting in gain of function with excessive inhibition of growth and differentiation. Curiously, loss-of-function mutations affecting the cyclin-dependent kinase-binding domain of this protein are associated with overgrowth syndromes, such as Beckwith-Wiedemann syndrome.
CHD7 (CHARGE Syndrome)
The CHARGE syndrome is associated with mutations in the chromodomain helicase DNA binding protein-7 (CHD7) gene. Features of this syndrome include eye coloboma, heart malformations, choanal atresia, short stature, genital anomalies, ear abnormalities, and hearing loss. Micropenis and cryptorchidism are found in males. Hypogonadotropic hypogonadism can occur. Although usually sporadic, autosomal dominant cases have been described. The CHD7 gene, located at chromosome 8q12.1-q12.2, codes for a large protein that participates in chromatin remodeling and transcription.
DHCR7 (Smith-Lemli-Opitz)
Several enzymes catalyze the conversion of lanosterol to cholesterol. Decreased activity of these enzymes leads to cholesterol deficiency. The enzyme, 7-dehydrocholesterol reductase, encoded by the 7-dehydrocholesterol reductase ( DHCR7 ) gene, located at chromosome 11q12-q13, catalyzes the last step in cholesterol biosynthesis. Smith-Lemli-Opitz (SLO) is an autosomal recessive disorder caused by DHCR7 loss-of-function mutations. As would be anticipated, elevated 7-dehydroxycholesterol concentrations are required to confirm the diagnosis.
Clinical features include urogenital anomalies, mental retardation, failure to thrive, facial abnormalities, developmental delay, behavioral abnormalities, and autism symptoms. The most common urogenital abnormalities include male-to-female sex reversal, hypospadias, and cryptorchidism. Facial abnormalities include microcephaly, broad nose, upturned nares, micrognathia, short neck, and cleft palate. Short thumbs, syndactyly of the second and third toes, and postaxial polydactyly are common. CNS malformations include septum pellucidum anomalies, agenesis of the corpus callosum, or holoprosencephaly in severe cases.
Infants often have hypotonia, feeding problems, and failure to thrive. Tube feedings may be necessary because of the poor weight gain. Impaired cholesterol biosynthesis may be associated with adrenal insufficiency. However, overt adrenal insufficiency is uncommon. Nevertheless, adrenal insufficiency, especially during stress, can occur. Thus the hypothalamic-pituitary-adrenal axis function should be assessed and glucocorticoid stress dosing may be beneficial. Although the efficacy remains unclear, dietary cholesterol supplementation is often prescribed.
DHCR7 mutations are associated with decreased cholesterol and accumulation of sterol intermediates proximal to the defective enzyme. Decreased cholesterol concentrations lead to decreased steroid concentrations, with subsequent decreases in glucocorticoid, mineralocorticoid, and sex steroid biosynthesis. In addition to serving as the precursor for vitamin D and bile acids, cholesterol is involved in SHH signaling and cell membranes. Available data indicate that accumulation of sterol intermediates rather than cholesterol deficiency leads to some of the clinical findings. Impaired SHH signaling contributes to malformations, such as agenesis of the corpus callosum, holoprosencephaly, and postaxial polydactyly. Using induced pluripotent stem cells from subjects with SLOS, data suggest that accumulation of 7-dehydrocholesterol and its isomer, 8-dehydrocholesterol, rather than cholesterol deficiency, likely contributes to the neurological manifestations.
Prenatal diagnosis can be performed by measurement of amniotic fluid 7-dehydrocholesterol concentrations. Women pregnant with affected fetuses have low plasma estriol; elevated blood 16α-hydroxyestrogens concentrations; and elevated urinary Δ7 and Δ8 unsaturated C 18 , C 19 , and C 21 dehydrosteroid concentrations, presumably because of impaired fetal cholesterol production. The incidence of biochemically confirmed SLO is estimated at 1/20,000 to 1/60,000 live births. The rate of heterozygosity for DHCR7 mutations is surprisingly high. Because the prevalence of SLO at 16 weeks of gestation is comparable to the prevalence at birth, early fetal loss and/or reduced fertility of carrier couples is likely occurring.
EMX2 Gene
One male patient presented with 46,XY DSD, a single kidney, and intellectual disability; he had a small microdeletion involving EMX2 .
GLI3 (Pallister-Hall Syndrome)
Pallister-Hall syndrome is characterized by genital anomalies, hypothalamic hamartoma, postaxial polydactyly, and imperforate anus. Hypospadias, micropenis, and bifid or hypoplastic scrotum are described in XY patients whereas hydrometrocolpos and/or vaginal atresia are described in XX patients. This autosomal dominant disorder is caused by mutations in the Gli-Kruppel Family Member 3 ( GLI3) gene, which is located at chromosome 7p14.1. The protein acts as both a transcriptional activator and a transcriptional repressor of downstream targets in the SHH pathway. Pallister-Hall is typically caused by frameshift/nonsense and splicing mutations located in the middle third of the gene, which are predicted to generate a truncated functional repressor protein. Curiously, mutations outside of this region are associated with Greigs cephalopolysyndactyly syndrome. Preclinical data using mouse knockout models showed aberrant ureteric budding, hydroureter, hydronephrosis, and renal hypoplasia.
HHAT (Nivelon-Nivelon-Mabille Syndrome)
The Nivelon-Nivelon-Mabille syndrome is an extremely rare disorder characterized by primordial short stature, generalized chondrodysplasia, microcephaly, brachydactyly, facial dysmorphism, and XY sex reversal. This syndrome is caused by autosomal recessive loss-of-function mutations in the O-acyl-transferase hedgehog acyl-transferase ( HHAT ) gene mapped to chromosome 1q32.2. The HHAT protein palmitoylates the N-terminal domain of the hedgehog proteins. The hedgehog proteins, SHH, DHH, and Indian hedgehog (IHH), play major roles in embryo patterning and differentiation. Based on the phenotype of the knockout mouse, loss of Hhat leads to testicular dysgenesis characterized by decreased testicular size, decreased testis cord, and almost complete absence of fetal Leydig cells. Curiously, the phenotype described for XY patients with DHH mutations and Dhh knockout mice resembles the phenotype reported for HHAT patients and Hhat knockout mice.
HOXA13 (Hand-Foot-Genital Syndrome)
HOX genes are a group of transcription factors involved in embryonic development and patterning. These genes are organized into clusters characterized by specific spatiotemporal expression patterns. The hand–foot–genital syndrome (HFGS) (OMIM #140000) is a rare, autosomal dominant disorder associated with mutations or deletions of the HOXA13 gene on chromosome 7p15. This syndrome is characterized by congenital genitourinary anomalies and variable skeletal anomalies. The skeletal anomalies mainly affect the hands and feet, such as short thumbs and halluces, shortening of the middle phalanges, fifth finger clinodactyly, and the fusion of distal and middle phalanges of the toes. Although approximately 50% of affected individuals have urogenital anomalies, many experience normal fertility. Males may have hypospadias or cryptorchidism. Females may have incomplete Mullerian fusion or bicornuate uterus associated with increased risks for spontaneous abortion, preterm delivery, or stillbirth. In some instances, the skeletal phenotype can be mild. Additional comorbidities include chronic urinary tract infections, ectopic ureteric orifices, chronic pyelonephritis, and renal failure.
KAT6B (Genitopatellar Syndrome)
The genitopatellar syndrome is a rare disorder characterized by skeletal dysplasia, flexion contractures, genital anomalies, craniofacial defects, and developmental delay associated with lysine acetyltransferase 6 ( KAT6B ) gene mutations. The skeletal features include hypoplastic or absent patellae, flat temporal bones, and brachydactyly. Genital anomalies include hypoplastic labia, clitoromegaly, scrotal hypoplasia, and cryptorchidism. Cardiac anomalies, hydronephrosis, and developmental delay have been described. The gene is located at chromosome 10q22.2 and contains 20 exons. The KAT6B protein is a component of the histone H3 acetyltransferase complex and functions as a histone acetyltransferase. The KAT6B protein functions as a chromatin modifier by catalyzing acetylation of specific lysines. Most mutations are de novo mutations. The Say-Barber-Biececker-Young-Simpson syndrome is also associated with KAT6B mutations; specific facial features consisting of blepharphimosis/ptosis and an immobile mask-like face distinguish these syndromes.
NR2F2 Gene
This gene encodes chicken oval albumin upstream promoter transcription factor (COUP-TF2), a nuclear receptor. This protein appears to function as a proovary and antitestis factor. Mutations in this gene are associated with a syndromic 46,XX DSD characterized by genital virilization, congenital heart disease, and varying somatic findings. Frameshift mutations have been reported in three 46,XX patients who were identified by genital ambiguity. All three patients had congenital heart disease. One infant died of hypoplastic left heart; no gonadal histology was available. The other two patients carried de novo c.97_103delCCGCCCG predicted to generate frameshift mutation, p.Pro33Alafs*77; they also had blepharophimosis ptosis epicanthus inversus syndromes (BPES) and uterus. One patient had ovotesticular disorder; gonadal histology was not available for the other patient.
Immunohistochemistry studies performed on human ovaries showed positive COUP-TF2 staining in FOXL2-negative stromal areas consistent with COUP-TF2 functioning as a proovary and antitestis factor in developing female gonads. Preclinical studies in mice showed that female embryos lacking Coup-tfII possess both Wolffian and Mullerian ducts; maintenance of the Wolffian ducts is androgen independent and is likely caused by enhanced phosphorylated extracellular signal-regulated kinase signaling in Wolffian duct epithelium. These data suggest that COUP-TFII actively promotes Wolffian duct regression in female embryos.
RSPO1
The R-spondin 1 (RSPO1 ) gene codes for a secreted furin-like domain protein that stabilizes β-catenin in the Wnt-signaling pathway. Mutations in this gene, located at chromosome 1p34, are associated with 46, XX sex reversal. This gene was initially identified by investigating in individuals with palmoplantar hyperkeratosis with squamous cell carcinoma of skin and sex reversal in one family. Affected XX, sex-reversed individuals lack Mullerian structures. A 46,XY individual, homozygous for RSPO1 mutations, fathered two children. An XX individual with ovotesticular disorder and palmoplantar keratoderma was found to be homozygous for a splicing mutation in the RSPO1 gene. In addition to sex reversal and palmoplantar keratosis, reported features include microphthalmia and nail dystrophy.
SAMD9 (MIRAGE Syndrome)
Mirage syndrome is characterized by myelodysplasia, infections, intrauterine growth restriction (IUGR), adrenal hypoplasia, XY male-to-female sex reversal, and enteropathy. Pulmonary disease, severe infections, and myelodysplasia contribute to deaths during the first 2 years of life. This disorder has been associated with heterozygous mutations in the sterile α motif domain–containing protein 9 ( SAMD9 ) gene located at chromosome 7q21.2. The 1589-amino-acid protein is highly expressed in the fetal adrenal; immunohistochemistry showed SAMD9 localization in the cytoplasm of adrenal definitive and fetal zone cells colocalized with cells positive for NR5A1 expression. SAMD9 is considered to be a tumor suppressor gene; mutations are gain-of-function mutations, leading to IUGR and consequences for adrenal gland and testes differentiation. Curiously, second mutations, such as acquired monosomy for chromosome 7, modify the phenotype and prolong survival. Buonocore and colleagues suggest that SAMD9 is essential to control the balance between cell proliferation and cell differentiation; activating SAMD9 mutations interfere with cell proliferation resulting in hypoplastic tissues.
SOX9
SOX9 is a member of the SRY-related HMG domain gene family located at chromosome 17q24.3-17q25.1 that encodes a 508-amino-acid protein. Heterozygous loss-of-function SOX9 mutations are associated with autosomal-dominant campomelic dwarfism and male-to-female sex reversal. Features of campomelic dwarfism include congenital bowing of long bones, hypoplastic scapulae, 11 pairs of ribs, narrow chest, congenital dislocated hips, and clubfeet. Facial features include micrognathia, cleft palate, large head, flat nasal bridge, and low-set malformed ears. When bone malformations are severe, postnatal survival may be limited. Affected 46,XY fetuses manifest sex reversal, with external genital differentiation, ranging from ambiguous to female. Gonadal dysgenesis and persistence of Mullerian duct derivatives are typical. Phenotypic heterogeneity with differing phenotypes, including ovotesticular DSD and complete sex reversal, has been described in affected siblings. Acampomelic dysplasia has been described in which the typical limb anomalies are absent. Patients with acampomelic dwarfism and sex reversal have been reported. Familial campomelic dysplasia associated with a deletion upstream of SOX9 has been described in a mother and 46,XY child with female external genitalia, normal uterus, and streak gonads.
SOX9 mutations can affect DNA-binding affinity, DNA bending ability, nuclear import, transactivation, and nuclear export. Haploinsufficiency is the mechanism responsible for many of the consequences of SOX9 mutations. Somatic cell mosaicism, de novo germline mutations, and mitotic gene conversion events have been described. Mutations located upstream of SOX9 in the regulatory regions have been identified in patients with varying degrees of sex reversal. Duplications of the eSR-A locus were associated with 46,XX testicular disorder and 46,XX ovotesticular disorder, whereas deletions have been identified in patients with 46,XY sex reversal.
TBX3 (Ulnar-Mammary syndrome)
The Ulnar-Mammary syndrome is characterized by cryptorchidism, micropenis, breast hypoplasia/aplasia, nipple hypoplasia/aplasia, delayed puberty, absent axillary hair, and skeletal anomalies. Skeletal anomalies include hypoplasia of humerus and ulna, oligodactyly, polydactyly, and finger anomalies. The TBX3 gene plays a role in development of the dorsoventral limb axis. Additional endocrine features include hypogonadotropic hypogonadism, short stature, growth hormone deficiency, and obesity. Neuroimaging studies have identified pituitary and brain malformations.
WT1
The Denys-Drash syndrome is characterized by genitourinary anomalies, Wilms tumor, nephropathy, and Wilms tumor suppressor (WT1) gene mutations. Among affected 46,XY individuals, the external genitalia can range from ambiguous to normal female. Internal genital differentiation varies because of inconsistent Wolffian and/or Mullerian structure development and regression. Typically, the nephropathy begins during the first few years of life, manifests with proteinuria, and results in end-stage renal failure because of focal or diffuse mesangial sclerosis. Typically, the gonads are usually dysgenetic in 46,XY individuals. Affected 46,XX individuals typically have normal female external genital development.
However, a novel WT1 mutation was reported in an XX individual identified at birth because of clitoromegaly and a single perineal opening. She was found to have an immature right uterine tube; gonadal histology revealed bilateral testes with seminiferous tubules containing predominantly Sertoli cells and rare germ cells. The karyotype of both gonads was XX. Genetic analysis revealed a de novo frameshift variant in exon 10, which encodes the fourth DNA finger. This variant is predicted to generate a deleterious mutation p.Arg485Glyfs*14, which appears to be a gain-of-function mutation increasing DNA binding affinity, promoting NR5A1 overexpression and SOX9 upregulation, and culminating in testicular development.
The Wilms tumor suppressor ( WT1 ) gene, located at chromosome 11p13, plays an important role in both kidney and gonadal differentiation. Through alternative splicing, multiple translation start sites, and posttranslational RNA editing, over 30 isoforms are derived from this one gene. The carboxyl terminal domain of the WT1 protein contains four zinc fingers that serve as the nucleic acid binding domain.
The two major isoforms differ by the inclusion or exclusion of three amino acids, lysine, threonine, and serine (KTS), between the third and fourth zinc-finger domains. Subnuclear localization studies have shown that the –KTS form colocalizes predominantly with transcription factors and preferentially binds to DNA, whereas the + KTS form colocalizes mainly with splicing factors and plays a role in RNA processing. The ratio of the + KTS/–KTS isoforms appears to be tightly regulated. Hence depending on cell context, WT1 can function as a transcriptional activator, a transcriptional repressor, or tumor suppressor. WT1 plays a role in the balance between mesenchymal-epithelial transitions.
Patients with WT1 mutations manifest phenotypic heterogeneity. Although Wilms tumor and genitourinary abnormalities can be associated with heterozygous WT1 deletions, only 6% to 15% of sporadic Wilms tumors are associated with WT1 mutations. Heterozygous deletions at chromosome 11p13 can be part of a contiguous gene deletion syndrome known as WAGR syndrome (Wilms tumor, aniridia, genitourinary anomalies, gonadoblastoma, and mental retardation). In general, missense mutations in exons 6–9 are associated with severe gonadal dysgenesis and early-onset nephropathy. The (–) KTS form appears to be protective from development of Wilms tumor, whereas mutations located in the N-terminal repression domain are associated with development of Wilms tumor.
Frasier syndrome is characterized by gonadal dysgenesis, progressive glomerulopathy, and an increased risk for gonadoblastoma. Wilms tumor is extremely rare in Frasier syndrome. The typical renal lesion is focal glomerular sclerosis. The majority of cases are associated with a specific WT1 point mutation in intron 9, associated with altered splicing and decreased amounts of the + KTS isoform.
Meacham syndrome is characterized by genital anomalies, cyanotic congenital heart defects, and pulmonary hypoplasia secondary to diaphragmatic abnormalities. In 46,XY infants, the spectrum of internal genital anomalies extends from presence of a uterus to a blind ending vaginal pouch. External genitalia have been described as ranging from normal female to ambiguous.
XH2 (ATRX Syndrome)
ATRX (α-thalassemia, mental retardation, X-linked protein) syndrome is an X-linked disorder characterized by mild alpha-thalassemia, severe mental retardation, and genital abnormalities because of mutations in the XH2 gene located at chromosome Xq13.3. Urogenital anomalies, including ambiguous genitalia, cryptorchidism, hypoplastic scrotum, hypospadias, shawl scrotum, and small penis occur in approximately 80% of patients. Additional features include short stature, psychomotor retardation, microcephaly, seizures, talipes equinovarus, and gastrointestinal problems. The facies are described as coarse with midface hypoplasia, short nose, and widely spaced incisors. The hemoglobin H inclusions associated with alpha-thalassemia can be demonstrated on brilliant cresyl blue stained peripheral blood smears. Typically, Wolffian duct structures are present, whereas Mullerian duct structures and germ cells are absent. Phenotype-genotype correlations are inconsistent. Most cases are inherited from carrier mothers and most, but not all, carrier females show preferential inactivation of the X chromosome carrying ATRX mutations.
This disorder, also known as Carpenter-Waziri syndrome, is caused by mutations in the ATRX (also known as XH2 or XHP ) gene located at Xq13.3. The ATRX protein is a member of the SWI/SNF DNA helicase family and consists of three domains: an N-terminal containing a plant-homeodomain-like (PHD) domain, a coiled-coil motif, and C-terminal helicase domain. The urogenital anomalies are typically associated with mutations located in the PHD domain and predicted to generate a truncated protein. During meiosis, ATRX maintains genomic integrity by modulating DNA damage response mechanisms. ATRX mutations are associated with abnormal methylation of repetitive DNA sequences, failure to establish necessary epigenetic modification for X chromosome inactivation, and impaired silencing of imprinted genes.
Sertoli cell ATRX knockout mice had small testes, decreased seminiferous tubule volume, decreased Sertoli cell number, and increased Sertoli cell apoptosis associated with prolonged G2/M phase during fetal life. This finding is consistent with the concept that ATRX protein expression impacts on G2-M progression with ultimate consequences on cell survival. Tissue-specific consequences of the epigenetic regulation and disrupted cell cycle progression may explain the disparate features associated with ATRX mutations.
Disorders associated with 46,XX disorders of sex development and 46,XY disorders of sex development ( Table 6.3 )
SF1/NR5A1 Gene
The SF1/NR5A1 gene, located at chromosome 9q33, codes for a 461-amino-acid protein. This protein is a nuclear receptor transcriptional regulator involved in adrenal, gonadal, and hypothalamic development and function. The protein contains an amino terminal DNA binding domain containing two zincfingers, a hinge domain, and ligand-binding domain. The protein modulates expression of SRY , SOX9 , AMH , and AMHR . In Leydig cells, NR5A1 promotes expression of LHCGR , STAR , CYP11A1 , and CYP17A1 , which are required for testosterone biosynthesis.
| Gene | Location | Phenotype | Genetic Defects | Mode of Inheritance |
|---|---|---|---|---|
| AMH | 19p13.3 | Persistent Mullerian duct syndrome, gonadal dysgenesis (OMIM #261550) | Point mutations | AR |
| AMHR2 | 12q13.13 | Persistent Mullerian duct syndrome (OMIM #261550) | Point mutations | AR |
| AR | Xq12 | Androgen resistance, gonadal dysgenesis (OMIM # 300068) | Deletions, point mutations | X-linked |
| AXIN1 | 16p13.3 | Cryptorchidism, caudal duplication anomaly (OMIM # 603816) | Point mutations | unknown |
| CBX2 | 17q25 | Complete gonadal dysgenesis, ovotesticular DSD (OMIM #613080) | Point mutations | AR |
| DHH | 12q13.12 | Gonadal dysgenesis, partial or complete (OMIM #233420) | Point mutations | AR |
| DMRT1 | 9p24.3 | Gonadal dysgenesis, partial or complete Additional anomalies in patients with 9p24.3 deletions (OMIM #154230) | Point mutations, 9p24.3 deletions (contiguous gene deletion syndrome) | AD |
| ESR2 | 14q23.2-q23.3 | XY sex reversal (OMIM # 601663) | Point mutations | AD, AR |
| FGFR1, multiple | 8p11.23, multiple | Hypogonadotropic hypogonadism with or without anosmia, see (OMIM # 147950) for additional loci | Deletions/duplications, point mutations | AD, AR, oligogenic |
| GATA4 | 8p23.1 | Testicular anomalies with/without congenital heart disease (OMIM# 615542 ) | Deletions/duplications, point mutations | AD |
| KAL1 | Xp22.31 | Hypogonadotropic hypogonadism with anosmia (OMIM #308700) | Deletions/duplications, point mutations | X-linked |
| LHCGR | 2p16.3 | Leydig cell hypoplasia (OMIM #238320) | Point mutations | AR |
| MAMLD1 | Xq28 | Micropenis, bifid scrotum, penoscrotal hypospadias (OMIM #300758) | Point mutations | X-linked |
| MAP3K1 | 5q11.2 | Gonadal dysgenesis (OMIM #613762) | Point mutations | AD |
| NR0B1(DAX1) | Xp21.2 | Complete gonadal dysgenesis, sex reversal (OMIM #300018) | Duplications, X-autosome translocations, deletions upstream | X-linked |
| NR5A1 (SF1) | 9q33.3 | Underandrogenization, complete gonadal dysgenesis, adrenal insufficiency (OMIM # 612965) | Deletions/duplications, point mutations | AD |
| SOX8 | 16p13.3 | Ambiguous genitalia, complete gonadal dysgenesis (PMID: 21373258 ) (28) | Point mutations, upstream duplications | AD |
| SRY | Yp11.2 | Gonadal dysgenesis (OMIM #400044) | Deletions/duplications, point mutations | Y-linked |
| WWOX | 16q23.1-q23.2 | Gonadal dysgenesis (PMID: 22071891 ) | Deletions | AD |
| ZFPM2 (FOG2) | 8q23.1 | Gonadal dysgenesis with/without congenital heart disease (OMIM #616067) | Point mutations | AR |
| ZNRF3 | 22q12.1 | Gonadal dysgenesis (OMIM# 612062) | Point mutations | AD |
Phenotypic heterogeneity is characteristic for patients with NR5A1 mutations. Among 46,XY individuals, reported phenotypes range from impaired testicular development to male infertility. Among 46,XX individuals, reported phenotypes range from 46,XX ovotesticular disorder to primary ovarian insufficiency. Adrenal insufficiency may be the primary feature in some patients. Nonsense, frameshift, and missense changes have been identified. Most mutations are heterozygous. Familial cases have been reported. Differing phenotypes have been described in a single family.
Because of the multiple NR5A1 actions in gonadal differentiation and function, several potential mechanisms of action are possible with NR5A1 mutations. Among 46,XY individuals, NR5A1 mutations cannot adequately activate the male pathway leading to undervirilization. Among XX (SRY-negative) virilized patients, based on in vitro studies, NR5A1 mutations do not appear to abnormally activate the male pathway. Rather, in vitro studies showed that p.Arg92Trp or p.Ala260Val missense mutations repressed Wnt signaling, thus interfering with ovarian differentiation in virilized 46,XX (SRY-negative) patients. In other words, downregulation of the female “antitestis” genes decreases inhibition of the male developmental pathway, shifting cell fate determination to promote testicular differentiation in XX individuals. Curiously, 46XX individuals with a different missense mutation, p.Arg92Gln, have been reported as having adrenal insufficiency and with or without ovotesticular DSD.
Incomplete penetrance and variable expression are recognized characteristics of NR5A1 mutations. The NR5A1 mutations illustrate how specific variants in a transcription factor act as a cell/organ switch to direct developmental outcomes. With whole exome sequencing (WES), variants in other genes have been demonstrated. Hence, oligogenicity or modifier genes may influence phenotypic expression for NR5A1 mutations.
SOX8
SOX8 is located at chromosome 16p.13.3. SOX8 mutations are associated with 46,XY sex reversal and infertility in both 46,XX and 46,XY individuals. Heterozygous SOX8 missense mutations were identified in individuals with 46,XY sex reversal.
Nonsyndromic 46,XY disorders of sex development (Gonadal dysgenesis) (see Table 6.3 )
Chromobox Homolog 2 ( CBX2 )
The CBX2 gene, mapped to chromosome 17q25, is a subunit of the chromatin-associated polycomb repressive complex 1. As noted earlier, the polycomb group proteins regulate chromatin structure, ultimately influencing gene expression. Two isoforms exist, CBX2.1 and the shorter CBX2.2 . Loss-of-function missense CBX2.1 mutations were identified in a 46,XY infant identified by a mismatch between prenatal karyotype and female external genital appearance at birth. Subsequent evaluation identified a normal uterus and ovarian-like tissue on histology. Two patients with CBX2.2 mutations have been reported; both patients had 46,XY karyotypes. One patient had dysgenetic testes, whereas no gonadal tissue could be identified in the second patient. Available data suggest that CBX2.1 is involved in testis determination, whereas CBX2.2 is involved in early gonad development. The factors that regulate alternative splicing of this gene are unknown.
Desert Hedgehog
The Desert hedgehog ( DHH ) gene is located on chromosome 12q12-q13.1 and encodes a protein consisting of 396 amino acids. DHH is a member of the Hedgehog family and is also expressed in the Schwann cells of the peripheral nervous system. This is an autosomal recessive disorder associated with XY gonadal dysgenesis and minifascicular neuropathy. However, neuropathy is an inconsistent feature. DHH promotes formation of the connective tissue sheath around peripheral nerves and is essential for their survival.
One 46,XY patient had female external genitalia, polyneuropathy, a testis on one side, and a streak gonad on the other side. Genetic analysis showed homozygosity for a single nucleotide substitution at the initiation codon, which is predicted to abolish initiation of translation at the normal start site. Histological analysis of the sural nerve revealed extensive formation of minifascicles within the endoneurium. The associated minifascicular neuropathy has been attributed to developmental malformation of peripheral nerves and is characterized by the presence of many small fascicles with glove and stocking-type sensory impairment. Gonadal tumors, including seminoma-in-situ, gonadoblastoma, and dysgerminoma have been reported.
Doublesex and Mab-3–Related Transcription Factor 1 ( DMRT1 ) and Doublesex and Mab-3–Related Transcription Factor 2 ( DMRT2 )
Monosomy for distal chromosome 9p has been reported in male-to-female sex reversal. Most deletions associated with sex reversal involve the chromosome region 9p24.3 where three DMRT genes are located. DMRT1 appears to be involved in Sertoli and cell differentiation. External genitalia have been described as ambiguous or female. The external genitalia may appear symmetric or asymmetric. Differentiation of internal genitalia is highly variable, with the presence of Mullerian and Wolffian remnants being reported. In addition to sex reversal, clinical features include mental retardation, low-set ears, trigonocephaly, wide nasal bridge, single palmar creases, heart defects, epilepsy, and scoliosis. Genital anomalies reported in XY individuals include gonadal dysgenesis, ovotestis, hypospadias, penoscrotal inversion, and cryptorchidism. Gonadoblastoma have been reported. Although phenotype/genotype correlations are not apparent, haploinsufficiency for DMRT1 appears to be sufficient to cause gonadal dysgenesis. Female external genitalia and short stature were described in a patient with mosaic 46,XY karyotype characterized by ring chromosome 9 and DMRT1 haploinsufficiency.
GATA4
The GATA proteins comprise a family of tissue-specific transcription factors that contain two conserved zinc-finger domains. The C-terminal zinc finger is required for DNA recognition and the N-terminal zinc finger contributes to stability; both zinc fingers are required for protein-protein interactions with other transcription factors. The GATA4 gene is mapped to chromosome 8p23.1. Heterozygous GATA4 mutations are associated with congenital heart disease and XY disorders of sex development. The observed phenotypic heterogeneity has been attributed to incomplete penetrance and oligogenicity. Three males in one family were reported to manifest congenital heart disease and varying abnormalities in male sex development, whereas the female carrier had congenital heart disease without an ovarian phenotype. This specific mutation, p.Gly221Arg, abrogated the ability of the GATA4 protein to interact with the ZFPM2/FOG2 protein and disrupted synergistic activation of the AMH promoter by GATA4 and NR5A1.
Mastermind-Like Domain Containing 1 ( MAMLD1 )
MAMLD1 was identified during studies investigating the genetic basis of X-linked myotubular myopathy. This gene is also known as chromosome X open reading frame 6 ( CXorf6 ). However, some variants have been detected in normal individuals. In vitro, most MAMLD1 variants acted similarly to the wild type with the exception of the L210X variant. The lack of a significant reproductive phenotype in transgenic knockout mice raises doubt whether MAMLD1 is sufficient to cause a DSD. Hence these findings suggest that MAMLD1 variations are insufficient to account for a patient’s DSD phenotype.
Mitogen-Activated Kinase Kinase Kinase 1 ( MAP3K1 )
The MAP3K1 gene is located at chromosome 5q11.2. This gene encodes a protein involved in MAPK signaling. Phenotypes of affected individuals include 46,XY complete gonadal dysgenesis, perineoscrotal hypospadias, cryptorchidism, and variable Mullerian duct development. Gonadoblastoma and dysgerminoma have been reported. Inheritance appears to be sex-limited autosomal dominant. Variations associated with gonadal dysgenesis increase phosphorylation of the downstream targets, MAPK11 (p38), MAP3K (ERK1), and MAPK1 (ERK2) and increase binding of the cofactors RHOA and MAP3K4 (AXIN1); the net result is decreased SOX9 expression. Hence these gain-of-function mutations repress the testis-promoting factors and stabilize beta-catenin, favoring the ovarian differentiation pathway.
Nuclear Receptor Subfamily 0, Group B, Member 1 ( NROB1 )
This two-exon gene, NROB1, located at Xp21.2 encodes DAX1. The 460-amino-acid protein is an orphan nuclear receptor lacking a typical zinc-finger DNA-binding domain. The N terminus of the 470-amino-acid protein contains a novel DNA-binding domain, whereas the C terminus shows characteristics of a nuclear hormone receptor ligand-binding domain. NROB1 is expressed throughout the HPG axis. The DAX1 protein functions as a transcriptional repressor of many genes, including NR5A1 and some steroidogenic enzyme genes.
Duplication of the DAX1/NROB1 locus is associated with male-to-female sex reversal. External genital differentiation ranges from female to ambiguous. Descriptions of internal genitalia include the presence of Mullerian and Wolffian structures. Gonads are typically described as streaks. Using high-resolution array comparative genome hybridization (CGH), a submicroscopic interstitial duplication of the DAX1 / NROB1 gene was discovered during thorough evaluation of two sisters. This 637-kb duplication included NROB1 , four MAGEB genes, CXorf21, glycerol kinase ( GK ), and part of the MAP3K7IP3 gene. The older sister had presented with primary amenorrhea, female external genitalia, 46,XY karyotype, and gonadal dysgenesis; the younger phenotypic female sister was prepubertal, but found to have 46,XY karyotype.
Loss-of-function mutations are associated with X-linked adrenal hypoplasia congenita (AHC). In this disorder, development of the fetal adrenal cortex is normal. However, the adult or definitive adrenal cortex fails to develop. Adrenal insufficiency may not be evident in the immediate neonatal period but may become obvious during early infancy. Although adrenal insufficiency generally manifests in infancy or early childhood, phenotypic heterogeneity in severity and age at presentation occurs. Unilateral or bilateral cryptorchidism can also occur. At the age of expected puberty, hypogonadotropic hypogonadism caused by hypothalamic and pituitary dysfunction may occur among affected males. Delayed puberty has been recognized in heterozygous females. One female homozygous for NROB1 mutations has been reported. Her phenotype was hypogonadotropic hypogonadism. As part of a contiguous gene deletion syndrome, X-linked AHC can be associated with glycerol kinase deficiency, Duchenne muscular dystrophy, ornithine transcarbamylase deficiency, and mental retardation.
Nonsense mutations have been identified throughout the gene. Missense mutations account for 20% of mutations associated with AHC. These mutations tend to cluster in the carboxyl terminal of the protein corresponding to the putative ligand-binding domain and impair the transcriptional repression activity of the protein. One missense point mutation, located in the hinge region of the protein, was identified in an 8-year-old girl with clinical and laboratory features indicative of adrenal insufficiency. Additional studies showed that this mutation hindered nuclear localization of the protein. Curiously, the hemizygous father and heterozygous younger sister of the proband did not manifest the AHC phenotype.
PBX1 (Pre-B-Cell-Leukemia Factor 1)
Pre-B-cell-leukemia factor 1 ( PBX1 ), located at chromosome 1q23.3, is a member of the Three Amino Acid Loop Extension (TALE) proteins. PBX1 dimerizes with other proteins to generate nuclear complexes that promote binding specificity of HOX proteins to DNA. Mutations in PBX1 have been reported among patients with congenital anomalies affecting the kidney and urinary tract, in association with developmental delay, ear anomalies, and congenital heart disease. A de novo PBX1 mutation, p.Arg235Gln, located within the conserved homeodomain, was identified in a 46,XY child with female external genitalia; the right gonad consisted of sparse seminiferous tubules with fibrous tissue and the left gonad consisted solely of fibrous tissue. Expression studies showed that this missense mutation failed to localize in the nucleus, implying PBX1 is necessary for testicular development and that this mutation is a loss-of-function mutation.
Protein Phosphatase Two Regulatory Subunit B”Gamma ( PPP2R3C )
This gene, mapped to chromosome 14q13.2, has 13 exons encoding a 453 amino acid protein. In conjunction with scaffolding A and catalytic C subunits, the three proteins comprise a heterotrimeric protein phosphatase 2A, which functions in the reversible dephosphorylation of phosphoproteins. Four phenotypic female patients with 46,XY gonadal dysgenesis, flat facies, bilateral epicanthal folds, low-set posteriorly rotated ears, and single palmar crease have been described. All four were homozygous for missense mutations; parents were heterozygous carriers. Curiously, three of the four heterozygous fathers were found to have impaired spermatogenesis characterized by teratozoospermia with severe head, acrosomal, and nuclear anomalies.
SRY
SRY is a single exon gene encoding a 204-amino-acid protein. The protein contains an HMG DNA-binding domain flanked by nuclear localization signals (NLS). The majority of sex-reversing SRY mutations are located in the HMG/NLS domain and affect DNA-binding affinity, DNA bending ability, or nuclear localization. Mutations located in the N-terminal and C-terminal domains have been identified in 46,XY sex reversed individuals. Mutations in the non-HMG domains may influence transcriptional activation, DNA binding, or protein-protein interactions. Paternal mosaicism for SRY mutations in which different cells carry different SRY genes has been described. More puzzling are the pedigrees in which fathers and unaffected brothers carry the identical mutant SRY allele as the propositus. These paradoxical findings implicate involvement of other genes, gene-gene interactions, and gene-environment interactions in the process of sexual differentiation. Nevertheless, only 15% to 20% of cases of 46,XY DSD because of gonadal dysgenesis can be attributed to SRY mutations. Mutations in the SRY gene have also been reported in patients with stigmata typical of Turner syndrome and 45,X/46XY karyotype.
WW Domain-Containing Oxidoreductase ( WWOX )
The WWOX gene is mapped to 16q23.1-16q23.2. This gene encodes a cytoplasmic protein that plays a role in growth, differentiation, and tumor suppression. One patient with 46,XY sex reversal was found to carry a heterozygous deletion of exons 6 to 8 (amino acids 173–352). This patient had unfused labioscrotal folds, bilateral nonpalpable gonads, clitoral hypertrophy, a single perineal opening, and intraabdominal dysgenetic gonads. WWOX mutations have been identified in autosomal recessive cerebellar ataxia with epilepsy and mental retardation.
Zinc Finger and Ring Finger Protein 3 ( ZNRF3 )
ZNRF3 mapped to chromosome 22q12.1 encodes a transmembrane E3 ubiquitin ligase that typically interferes with WNT signaling in the gonad to promote testicular differentiation. Heterozygous mutations have been reported in 5 XY patients with sex-reversal; four of five patients presented in late adolescence with delayed puberty.
Zinc Finger Protein, Multitype 2 ( ZFPM2 )
The Friend of GATA 2 (FOG2) protein, encoded by ZFPM2 and mapped to chromosome 8q23.1, is a transcriptional cofactor that modulates the activity of GATA4 by binding to its N-terminal zinc finger. One boy with a balanced translocation and breakpoint with intron 4 of the FOG2 gene had congenital heart disease, retractile testes, and hypergonadotropic hypogonadism.
Ovotesticular disorder of sex development
Ovotesticular DSD is defined as the presence of ovarian tissue with follicles and testicular tissue with seminiferous tubules in the same individual. External genital appearance does not predict gonadal histology. The gonads can be an ovotestis, ovary, and/or testis. The most common histology is an ovotestis. An ovotestis is typically located on the right side and is often intraabdominal. An ovotestis can appear ovoid or bilobed on gross appearance. In most ovotestes, ovarian and testicular tissue show distinct separation in an end-to-end arrangement. In this situation, the testicular zone typically shows poor differentiation of the tunica albuginea with atypical interstitial tissue. In some instances, oocytes are interspersed among seminiferous cords/tubules. The proportion of ovarian and testicular tissue differs among patients; uterine development also varies.
The most common karyotype is 46,XX. Mosaic karyotypes, such as 46,XX/46,XY, 46,XX/47,XXY, 45,X/46,XY, 47,XYY/46, and XY/45,X have been described. In some cases, the amount of Y chromosomal material in peripheral blood lymphocytes is limited, such as the SRY gene can be detected only by polymerase chain reaction (PCR) amplification. In other instances, SRY was apparently absent in peripheral blood lymphocytes and detected in testicular tissue. A deletion of the SRY promoter region was identified in the testicular tissue of a 46,XX patient with ovotesticular disorder. However, ovotesticular DSD in the absence of Y chromosomal material occurs. As noted elsewhere in this chapter, specific mutations, for example, NR5A1 , NR2F2 , RSPO1 , and SOX9, have been identified among patients with ovotesticular disorder.
Most cases are sporadic. Nevertheless, pedigrees in which both XX males and XX individuals with ovotesticular disorder coexist have been described. Although most patients present in infancy or childhood, phenotypic males can present with bilateral gynecomastia. One patient with 46,XX/46,XY karyotype presented in infancy with penoscrotal hypospadias, left descended gonad, and right undescended gonad; biopsies revealed normal prepubertal testis on the left and normal ovarian tissue on the right. Additional evaluation and management of this patient led to male sex of rearing accompanied by right salpingo-oophorectomy and hysterectomy. The patient subsequently presented at age 17 years with a painless scrotal mass, which was excised and found to be normal ovarian tissue. This case illustrates the potential for inconsistency in gonadal development among patients with ovotesticular disorder with the possibility of sampling errors in gonadal biopsies.
Limited outcome data regarding fertility are available. Among 33 patients followed longitudinally, germ cells identified in the testicular tissue during infancy degenerated, resulting in azoospermia. Histological findings among adults describe a dysgenetic Sertoli-only appearance with hyalinization of seminiferous tubules. Nevertheless, assisted reproductive techniques have been successfully used to achieve paternity in men with ovotesticular disorder. Several pregnancies have been reported among women with ovotesticular disorder.
Nonsyndromic XX disorders of sex development ( Table 6.4 )
46,XX Testicular Disorder of Sex Development
Testicular DSD is characterized by a male phenotype with a 46,XX karyotype. The frequency of the XX male syndrome is approximately 1 in 25,000 males. This form of DSD can be subclassified as SRY -positive and SRY -negative groups. The SRY-positive 46,XX males generally have normal male external genitalia, small azoospermic testes, hypergonadotropic hypogonadism, and often present with infertility. In most instances, the SRY gene is located on an X chromosome because of recombination between the X and Y chromosomes. However, translocation to an autosome can occur. One example was the incidental finding small testes, azoospermia, and translocation of the SRY gene onto the terminal end of chromosome 16q in a 61-year-old 46,XX man.
| Gene | Location | Phenotype | Genetic Defects | Mode of Inheritance |
|---|---|---|---|---|
| BMP15 | Xp11.22 | Ovarian dysgenesis a , primary amenorrhea (OMIM #300510) | Point mutations (paternally transmitted), deletions | X-linked |
| DCAF17 | 2q31.1 | Hypogonadism, ovarian dysgenesis, partial alopecia, Woodhouse-Sakati Syndrome (OMIM #241080) | Point mutations | AR |
| DIAPH2 | Xq21.33 | Primary or secondary amenorrhea (OMIM #300511) | Point mutations, deletions | X-linked |
| EIF2B1, EIF2B2, EIF2B3, EIF2B4, EIF2B5 | 12q24.31, 14q24.3, 1p34.1, 2p23.3, 3q27.1 | Leukoencephalopathy with vanishing white matter, gonadal dysgenesis, primary or secondary amenorrhea (OMIM #603896) | Point mutations | AR |
| FIGLA | 2p13.3 | Ovarian insufficiency (OMIM #612310) | Point mutations | AD |
| FMR1 | X | Fragile X premutation (OMIM #311360/#300869) | Trinucleotide expansion | X-linked |
| FOXL2 | 3q22.3 | Blepharophimosis with ovarian dysgenesis (OMIM # 110100) | Point mutations, translocations, deletion/duplications | AD |
| HFM1 | 1p22.2 | Ovarian dysgenesis, ovarian insufficiency (PMID: 27603904 ) (29) | Point mutations | unknown |
| LMNA | 1q22 | Ovarian dysgenesis with cardiomyopathy (OMIM # 212112 ) | Point mutations | AR |
| MCM8 | 20p12.3 | Ovarian dysgenesis, primary amenorrhea (OMIM #236700) | Point mutations | AR |
| MCM9 | 6q22.31 | Ovarian dysgenesis, primary amenorrhea (OMIM #616185) | Point mutations | AR |
| MKKS | 20p12.2 | McKusick-Kaufman syndrome hypogenitalism, hydrometrocolpos (OMIM #236700) | Point mutations | AR |
| NOBOX | 7q35 | Ovarian dysgenesis, primary ovarian insufficiency (OMIM #611548) | Point mutations | AD & AR |
| NR5A1 (SF1) | 9q33.3 | Gonadal dysgenesis, ovotesticular DSD, secondary ovarian insufficiency, sex reversal (OMIM #612964) | Deletions/duplications Point mutations | AD |
| NUP107 | 12q15 | Ovarian dysgenesis, primary amenorrhea (OMIM #607617) | Point mutations | AR |
| PMM2 | 16p13.2 | Ovarian dysgenesis, cerebellar atrophy, mental impairment, pigmentary retinopathy (OMIM #212065) | Point mutations | AR |
| POF1B | Ovarian insufficiency (OMIM #300604) | |||
| PSMC3IP | 17q21.2 | Ovarian dysgenesis, primary amenorrhea (OMIM #614324) | Point mutations | AR |
| REC8 | 14q12 | Ovarian dysgenesis, ovarian insufficiency (PMID: 27603904 ) (29) | Point mutations | unknown |
| SMC1B | 22q12.31 | Ovarian dysgenesis, ovarian insufficiency (PMID: 27603904 ) (29) | Point mutations | unknown |
| SOHLH1 | 9q34.3 | Ovarian dysgenesis, ovarian insufficiency (PMID: 27603904 ) (29) | Point mutations | AR |
| SOHLH2 | Ovarian dysgenesis, ovarian insufficiency (OMIM #616066) | AD | ||
| SOX3 | Xq26.3 | Gonadal dysgenesis, virilizing 46,XX sex reversal (OMIM # 300833 ) | Deletions/duplications | X-linked |
| SOX8 | 16p13.3 | Ovarian dysgenesis, primary or secondary amenorrhea | Point mutations | AD |
| SOX9 | 17q24.3-q25.1 | 46,XX sex reversal, virilizing chromosome 17q24 duplication syndrome (OMIM #278850) | Duplication 584–516 kb upstream of SOX9 | AD |
| SOX10 | 22q11.2q13 | 46,XX sex reversal, Waardenburg syndrome, peripheral demyelinating neuropathy | Duplication | AD |
| SPIDR | 8q11.21 | Ovarian dysgenesis, primary or secondary amenorrhea (OMIM # 615384) | Point mutations | AR |
| STAG3 | 7q22.1 | Ovarian dysgenesis, ovarian insufficiency (OMIM # 615723 ) | Point mutations | AR |
| SYCE1 | 10q26.3 | Ovarian dysgenesis, ovarian insufficiency (PMID: 27603904 ) (29) | Point mutations | unknown |
| WNT4 | 1p34.12 | Mullerian aplasia, hyperandrogenism, primary amenorrhea (OMIM # 158330 ) | Point mutations | AD |
a Also associated with large X chromosome deletions/duplications.
Approximately 10% of these patients are SRY -negative. The phenotypic spectrum ranges from genital ambiguity to normal male external genitalia. Testes are typically small because of meiotic arrest and germ cell degeneration. Molecular etiologies are more diverse for the SRY -negative 46,XX male. Duplication of SOX9 was described in one family; all affected family members had normal male secondary sexual characteristics and azoospermia. Duplications approximately 600 kb upstream of SOX9 have been identified in several SRY -negative XX males. Overexpression of the SOX10 gene at 22q13 was found in one patient with 46,XX sex reversal in association with multiple congenital anomalies.
SOX3 is a single exon gene mapped to Xq27. It plays a role in brain, pituitary, and craniofacial development. An SRY -negative 46,XX patient referred for evaluation of hypospadias and bilateral cryptorchidism was found to have bilateral ovotestes and SOX3 duplication. Several other patients have been reported with SOX3 duplications. Overexpression of SOX3 in a preclinical transgenic XX mouse model resulted in testicular development and suggested the hypothesis that SOX3 acts as a surrogate for SRY in this situation. Hypoplasia of the anterior pituitary and ectopic position of the posterior pituitary has been described in two patients.
XX Disorder of sex development/Premature ovarian failure
46,XX gonadal dysgenesis is a rare disorder associated with delayed puberty and premature menopause associated with hypergonadotropic hypogonadism (see Table 6.4 ). Affected individuals typically present with lack of spontaneous pubertal development, primary amenorrhea, and uterine hypoplasia.
As noted earlier, ovarian development is an active process involving multiple genes. However, mutations of genes involved in ovarian development are not typically associated with ambiguous genitalia; external genital development is typically female. Rather, these disorders typically present with delayed puberty, primary amenorrhea, and POI. In these disorders, follicles may fail to develop or may undergo premature atresia. Mutations in genes involved in ovarian development such as Forkhead box L2 ( FOXL2) , Newborn ovary homeobox ( NOBOX) , bone morphogenetic protein 15 ( BMP15) , and Factor in germline alpha (FIGLA) have been described. BMP15, a member of the TGF- β superfamily, influences granulosa cell growth and differentiation. Heterozygous missense BMP15 mutations have been identified among women with POI and associated with X-linked XX gonadal dysgenesis. Ovarian leukodystrophy is a leukoencephalopathy characterized by vanishing white matter and ovarian dysgenesis associated with mutations in the EIF2B2 , EIF2B4 , and EIF2B5 genes. A mutation in the proteasome 26 subunit, adenosine triphosphatase (ATPase), 3-interacting protein (PSMC3IP) gene has been described in autosomal recessive 46,XX gonadal dysgenesis; this loss-of-function mutation reduces estrogen-induced transcriptional activation of the protein encoded by this gene. Fragile X syndrome is caused by inactivation of the FMR1 gene caused by DNA hypermethylation of the promoter and expanded 5’-UTR CGG repeat region; the premutation allele containing 55-200 CGG repeats is associated with POI. Prevalence of premutation alleles in the general population is estimated at 1 per 150 to 300 females.
FOXL2
Forkhead box L2 (FOXL2) is a member of the winged helix/forkhead transcription family. The protein contains a DNA-binding domain, which is primarily located in the nucleus. The protein contains three α-helices. Mutations in FOXL2 , located at chromosome 3q23, are associated with the autosomal dominant BPES. BPES is characterized by eyelid dysplasia consisting of small palpebral fissures (blepharophimosis), ptosis, epicanthus inversus, and a broad nasal bridge. The phenotype of BPES type 1 consists of eye findings and premature ovarian failure. Patients with BPES type 2 manifest only the eye features. Patients heterozygous for FOXL2 mutations generally experience spontaneous pubertal development culminating in menarche, but undergo premature follicle depletion, leading to premature ovarian failure. The histological appearance of ovaries in patients with BPES type 1 is reported to vary from the presence of primordial follicles with some atretic follicles to the complete absence of follicles. Mutations generating nonsense mutations, predicted to generate truncated proteins lacking the transrepression domain, are associated with BPES type 1. Expansion of the polyalanine tract is associated with BPES type 2.
One major role of FOXL2 in the ovary appears to be transrepression of genes involved in steroidogenesis, such as StAR, P450scc, and aromatase to prevent premature differentiation and proliferation of granulosa cells. Loss of the transrepressor activity because of nonsense mutations likely contributes to the premature depletion of ovarian follicles and premature ovarian failure. One patient with ovotesticular disorder was found to have expression of both FOXL2 and SOX9 . However, in general, expression of these proteins is mutually exclusive.
Curiously in goats mutations in this locus are responsible for the autosomal dominant phenotype characterized by the absence of horns in male and female goats (polledness) and XX female-to-male sex reversal in a recessive manner.
NOBOX
The Newborn Ovary Homeobox ( NOBOX ) gene is expressed in germ cells and primordial oocytes. NOBOX encodes a basic helix-loop-helix transcription factor involved in the transition from primordial to primary follicles. The protein is expressed in fetal and adult ovaries. The NOBOX gene is located at chromosome 7q35. The phenotype of patients with NOBOX mutations includes delayed puberty, with primary amenorrhea and secondary amenorrhea. POI associated with NOBOX mutations shows both autosomal dominant and autosomal recessive inheritance patterns.
FIGLA
Factor in germline alpha ( FIGLA ) is a germ cell–specific transcription factor located at chromosome 2p13.3. FIGLA is expressed in the human fetal ovary. The phenotype of women with FIGLA mutations includes secondary amenorrhea and ovaries devoid of follicles. Both autosomal dominant and autosomal recessive inheritance have been described.
SPIDR
Scaffolding protein involved in DNA repair (SPIDR) , mapped to chromosome 8q11.21, encodes a protein involved in homologous DNA repair. Sisters who presented with primary amenorrhea were found to have hypergonadotropic hypogonadism associated with homozygous mutation.
NUP107
Nucleoporins comprise nuclear pore complexes that serve as the gatekeepers of nucleocytoplasmic transport. One such protein is nucleoporin-107, encoded by NUP107, is mapped to chromosome 12q15. Missense variants in NUP107 segregated with delayed puberty and hypergonadotropic hypogonadism associated with POI in two ethnically different families. Affected females were otherwise healthy; no male phenotype was evident.
WNT4 Gene
WNT4 is a secreted molecule that binds to members of the frizzled family of receptors, resulting in transcriptional regulation of target genes. WNT4 increases follistatin expression, which inhibits formation of the coelomic vessel (antitestis action) and supports ovarian germ cell survival (proovarian action). Duplication of the WNT4 gene, located at chromosome 1p31-1p35, has been associated with 46,XY male-to-female sex reversal. One such patient presented with ambiguous external genitalia accompanied by severe hypospadias, fibrous gonads, remnants of both Mullerian and Wolffian structures, cleft lip, microcephaly, and IUGR. A female-to-male sex reversed patient with kidney, adrenal, and lung dysgenesis was found to have a homozygous loss-of-function WNT4 mutation. Loss-of-function WNT4 mutations have been detected in 46, XX women with primary amenorrhea, secondary to Mullerian duct abnormalities and androgen excess. The phenotypes of these patients support the hypothesis that WNT4 plays a role in ovarian differentiation.
Vanishing testes
The terms testicular regression syndrome and vanishing testes are used to describe testicular absence in boys with undescended testes. In some instances, this situation is associated with ambiguous genitalia and undervirilization, which presumably represents regression of testicular tissue occurring between 8 and 14 weeks of gestation. Physical findings reflect duration of testicular function. At operation, a rudimentary spermatic cord and nubbin of testicular tissue may be identified. Histological examination of the testicular nubbins often reveals hemosiderin-laden macrophages and dystrophic calcification. One potential mechanism involves an antenatal vascular accident, associated with antenatal testicular torsion, leading to testicular regression. Another possibility is anomalous vascular development. Although usually sporadic, familial testicular regression has been described.
Disorders of cholesterol and steroid biosynthesis
Abnormalities affecting the biosynthetic pathways involved in cholesterol, cortisol, and sex steroid biosynthesis can lead to genital ambiguity ( Table 6.5 ). Steroidogenesis refers to the multiple enzyme-mediated processes through which cholesterol is converted to biologically active steroid hormones ( Fig. 6.2 ). This process depends on the coordinated regulation of cholesterol uptake into cells, transfer into mitochondria, and actions of tissue-specific enzymes. Details regarding the complexities of steroidogenic processes continue to unfold. Three pathways have been characterized: (1) canonical/classical, (2) “alternative backdoor,” and (3) 11-oxygenated C-19 androgens.
| Gene | Location | Phenotype | Genetic Defects | Mode of Inheritance |
|---|---|---|---|---|
| AKR1C2 and AKR1C4 | 10p15.1 | Complete gonadal dysgenesis, 3α-Hydroxysteroid Dehydrogenase Isozyme Deficiency (OMIM #614279) | Point mutations | AR |
| CYP5A | 18q22.3 | Undervirilized, delayed puberty methemoglobinemia (OMIM # 250790) | AR | |
| CYP11A1 | 15q24.1 | Adrenal insufficiency, sex reversal, partial or complete (OMIM # 613743 ) | Point mutations | AR/AD?, oligogenic |
| CYP11B1 | 8q24.3 | Virilizing congenital adrenal 11β-hydroxylase deficiency, hypertension (OMIM #202010) | Point mutations, deletions | AR |
| CYP17A1 | 10q24.32 | 17α-hydroxylase/17,20-lyase deficiency, genital ambiguity, delayed puberty (OMIM#202110) | AR | |
| CYP19A1 | 15q21 | Virilizing congenital adrenal placental aromatase deficiency (OMIM # 613546) | Point mutations | AR |
| CYP21A2 | 6p21.33 | Virilizing congenital adrenal hyperplasia caused by 21-hydroxylase deficiency (OMIM #201910) | Deletions, point mutations | AR |
| HSD3B2 | 1p12 | 3β-Hydroxysteroid dehydrogenase deficiency, genital ambiguity (OMIM #201810) | Point mutations | AR |
| HSD17B3 | 9q22.32 | 17β-Hydroxysteroid dehydrogenase deficiency, complete gonadal dysgenesis or hypovirilization and gynecomastia (OMIM# 264300) | Point mutations | AR |
| POR | 7q11.23 | virilizing congenital P450 oxidoreductase deficiency, (OMIM #613571) | Point mutations, deletions | AR |
| SRD5A2 | 2p23.1 | Ambiguous genitalia, 5α-reductase deficiency, (OMIM# 264600) | Point mutations, intragenic deletions | AR |
| STAR | 8p11.23 | Lipoid congenital adrenal hyperplasia (OMIM #201710) | Point mutations | AR |
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


