Fig. 19.1
Hormonal control of sexual differentiation
By 7 weeks of pregnancy a fetus has a dual system—Wolffian ducts with the potential for the development of a male and Müllerian ducts with the potential for the development of a female.
The Müllerian ducts regress in the presence of the testis while the Wolffian ducts develops in the epididymis, vas deferens, seminal vesicles, and ejaculatory ducts.
In the absence of testicles, the Wolffian ducts involute and the Müllerian ducts develop in the fallopian tubes, uterus, and upper third of the vagina [2].
Urogenital Sinus Development
Under the influence of androgens the urogenital sinus narrows to form the posterior urethra. The urogenital sinus forms the prostate gland and the Cowper’s bulbourethral glands.
In the absence of androgens the urogenital sinus differentiates in the lower two-thirds of the vagina and urethra, developing Skene’s paraurethral glands and Bartholin’s vestibular glands [2].
External Genitalia Development
In contrast to the internal genital, external genitalia stems from a common anlage, a midline genital tubercle formed in the eighth gestational week with urethral folds surrounded by side labioscrotal swellings.
The genital tubercle develops to form the glans penis in males and the clitoris in females.
The urethral folds develop forming the corpus spongiosum in males and the labia minora in females.
Labioscrotal swellings fuse in the midline to form the scrotum and ventral penis in males and remain separated to form the labia majora in females [2].
Hormonal Control of Sexual Differentiation (Fig. 19.1)
The fetus has an inherent tendency towards female differentiation. In the absence of gonads, the fetus will develop internal and external normal female genitalia. In contrast normal male differentiation requires intact testis (with Leydig cells and Sertoli functioning) and the 5α-reductase type 2 enzyme [3].
Male differentiation occurs as follows:
Wolffian ducts development, under the influence of testosterone secreted by Leydig cells.
Müllerian ducts regress, under the influence of a peptide produced by Sertoli cells, the anti-Müllerian hormone (AMH).
Development of the urogenital sinus under the control of dihydrotestosterone (DHT), synthesized from testosterone in tissues containing the 5α-reductase type 2 enzyme.
The following keys points to diagnosis should be considered:
The testis has a paracrine effect on adjacent tissue so that a testicle results in the Wolffian development and Müllerian regression, while a typical gonad may be associated with Wolffian regression and Müllerian development. This situation can be found with mosaicism 45X/46XY (often called mixed gonadal dysgenesis).
The presence of the uterus in a newborn with ambiguous genitalia indicates the presence of an ovary or the absence of gonadal tissue, or if there, testicular tissue should be dysgenetic with poor function of Sertoli cells.
46XY individuals with partial or complete androgen resistance secrete and respond normally to AMH, and no development occurs in the fallopian tubes, uterus, and upper third of the vagina.
Genetic Control of Gonadal Determination and Sexual Differentiation
Gonadal determination results from interplay between the SRY gene and others genes (e.g., SOX9, SF1, WT1, GATA4). The AMH gene (19p chromosome) controls the secretion of AMH by Sertoli cells. AMH has paracrine action on the AMH type II receptor (12q). Both testosterone and DHT mediate their effects via the androgen receptor (AR) (Xq11-q12), which is a transcription factor. Testicular descent is mediated by the secretion of certain factors such as insulin-like 3 (INSL3) and the G-protein coupled receptor (GREAT) [3].
Figure 19.2 summarizes the main genes involved in gonadal determination and sexual differentiation.
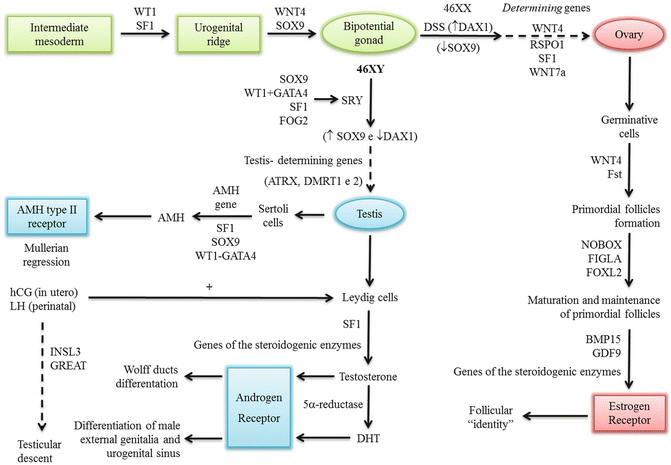
Fig. 19.2
Genes involved in sexual determination and differentiation
Mutations or deletions in the genes have been reported or proposed as causes of disorder of sexual development (DSD) or gonadal failure in humans.
Workup
The initial evaluation of a child with DSD includes medical history, family history, physical examination, hormonal secretion assessment, evaluation of internal anatomy, evaluation of ducts by imaging studies, and establishment of genetic sex by karyotype [4].
The clinical presentation is highly variable. Therefore, initially it is important to recognize when a child is suffering from DSD based on the following conditions [4]:
Obvious ambiguous genitalia.
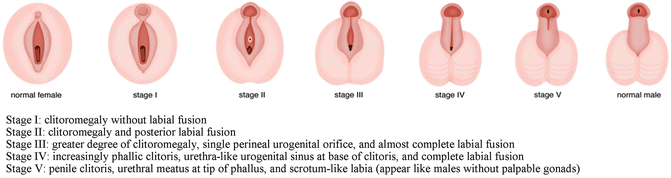
Apparently female genitalia with increasing clitoral (more than 6 mm in diameter or more than 9 mm in length), with labial fusion posterior fusion or inguinal/labial mass [5, 6].
Apparently male genitalia with bilateral cryptorchidism, micropenis (penile length less than −2.5 standard deviations from the mean for child’s age or in a term infant, the penile length is <2.5 cm, and penile diameter is <0.9 cm), isolated perineal hypospadias or mild hypospadias with cryptorchidism [5].
Discordance between phenotype and prenatal karyotyping.
The medical history should include [4]:
Family history of consanguinity, genital ambiguity, primary amenorrhea, or infertility
History of perinatal dehydration and/or hypoglycemia and/or death in the family suggests Congenital Adrenal Hyperplasia (CAH) salt wasting.
Medication virilizing or feminizing during pregnancy (especially in the first trimester) or maternal virilization (suggests a tumor producing androgens by the mother, P450-oxidoreductase deficiency, or presence of placental aromatase deficiency).
The general physical examination should include:
Features suggestive of malformation syndromes
Hydration, blood pressure
Genital examination:
Size and differentiation of the phallus
Location, size, and consistency of the gonads
Position of meatus
Genital skin pigmentation (hyperpigmentation suggests increased adrenocorticotropic hormone), which occurs in CAH.
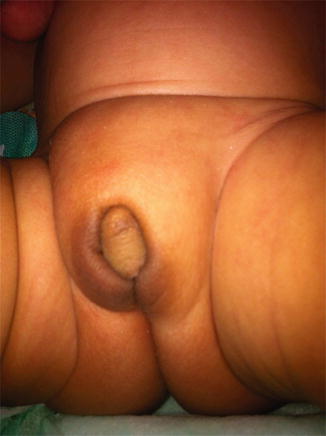
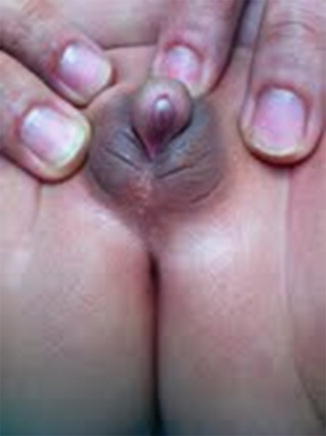
Hormonal Assessment
The following hormones should be evaluated on ambiguous genitalia, with the majority being used to diagnose CAH (Fig. 19.6):
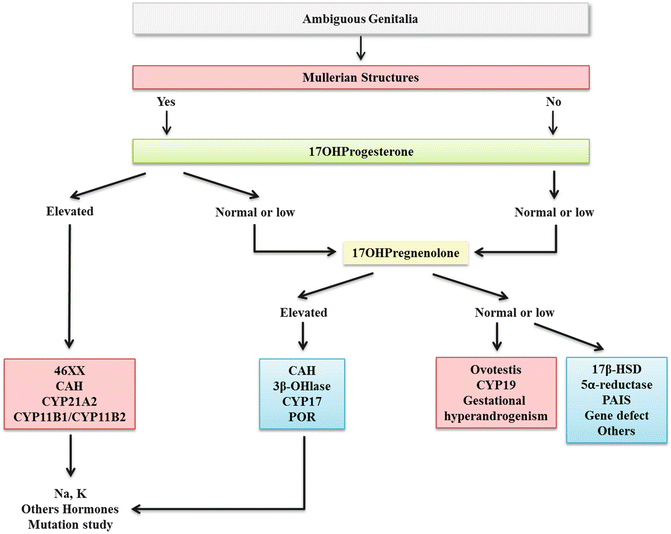
Fig. 19.6
Initial Approach to Ambiguous Genitalia
17OHprogesterone (17OHP)—Evaluate on the second day of life. High levels are useful to diagnosis CAH, CYP21A2, and CYP11B1 defects. Low levels are useful to diagnosis 17αhydroxylase/17lyase.
Pregnenolone/17OHpregnenolone—Defects of 3β − hydroxysteroid dehydrogenase and 17α − hydroxylase/17lyase, respectively.
Dehydroepiandrosterone (DHEA)—Defect of 17α − hydroxylase/17lyase.
The following tests are useful to evaluate testes function:
AMH—Low or undetectable in gonadal dysgenesis (46XY) and in 5α − reductase defect. Normal or high in androgen insensitivity, LH receptor, and steroidogenic protein defect [7].
Human chorionic gonadotropin (hCG) test—hCG has a LH-like function and stimulates testosterone synthesis in testes. There are various protocols for hCG stimulation, but intramuscular hCG 1,000–1,500 UI on 3 consecutive days is the most used. Testosterone levels should be done at basal and 24 h after the last dose. Normal response is considered twice the testosterone levels [8].
Imaging Studies
Ultrasonography (US), genitography, and magnetic resonance imaging (MRI) may be performed if deemed necessary [9].
US is useful to evaluate the presence or absence of gonads and Müllerian derivatives.
Genitography shows the presence or absence of the vagina, its relationship to the urethra, the level of the external sphincter, and cervical impression. It is helpful to establish the presence and size of the vagina or utricular pouch.
MRI is more sensitive than US in the evaluation of the gonads and had shown up to 100 % accuracy in detection of the testes [10]. MRI could be reserved for gonad identification when ultrasound examination fails. MRI and genitography are essential for proper pre-corrective surgery assessment.
Karyotype
Karyotype is performed, usually using peripheral leukocytes and the result will be 46XX, 46XY, 45X or mosaicism.
Gonadal Disorders
Ovotesticular DSD is a rare condition (4–10 % of cases of DSD). It refers to the presence of both ovarian and testicular tissue in various combinations in the same individual. This condition has been termed “true hermaphroditism” in the past, and these individuals present mixed ovarian and testicular tissue (either ovotestis, or ovary and testis). Often, the differentiation of internal genitalia coincides with the gonad on the ipsilateral side. The most common karyotype is 46XX, but there are also patients with 46XY or 46XX/46XY [11].
Gonadal Dysgenesis
In gonadal dysgenesis, a dysgenetic testis or a “streak gonad” replaced the gonad. There are two types: Pure or complete, and partial or mixed.
Pure gonadal dysgenesis: The neonate will have streak gonads and seems female but puberty will be delayed and the child will present with amenorrhea. The karyotype may be 46XX, 46XY, or a Turner syndrome karyotype (45X) [10]. Included in this group is Swyer syndrome with external female genitalia, uterus and fallopian tubes and karyotype 46XY. The cause may be a mutation in the SRY gene, although in the majority of cases, the etiology is unknown [14, 15].
Partial or mixed gonadal dysgenesis: There is a streak gonad in one side and a dysgenetic testis on the contralateral side. The genotype is 45X/46XY or 46XY. The phenotype depends on the amount of testosterone produced by the dysgenetic testis [16].
Gonadal Dysgenesis Associated with a Syndrome Phenotype
The mixed sex chromosome also includes 47XXX (Klinefelter syndrome), 45X (Turner Syndrome), and mosaicisms with normal genitalia.
Embryonic Testicular Regression Syndrome or Vanishing Testes
The testicular tissue regression occurs when the testes are lost during the phase of male differentiation. The penis and scrotum will form and continue to develop, but both testicles will be absent. The phenotype reflects the amount of testosterone produced before the regression.
Others
A 46XX DSD with evidence of functioning testicular tissue is important to evaluate the presence of genes of the Y chromosome, as it may be caused by translocation of the sex determining region of the Y (SRY) gene [17] or SOX9, SF1, WT1, DHH genes [18]. The diagnosis of SRY translocation can be diagnosed using a fluorescence in situ hybridization (FISH) probe for the SRY gene, and for the SOX9 duplication can be confirmed with a SOX9 FISH probe. These tests are available for either research or clinical evaluation.
Other Genetic Defects
Steroidogenesis Factor 1 (SF-1)—agonadism, adrenal hypoplasia with adrenal insufficiency, cryptorchidism, and micropenis.
Tumor Wilms gene (WT-1)—a transcriptional factor involving gonadal and renal development:
WAGR Syndrome—Wilms tumor, aniridia, genitourinary anomalies
Denys–Drash Syndrome—A triad: 46XY karyotype, undervirilization, and Wilms tumor.
Frasier Syndrome—46XY karyotype, gonadal dysgenesis with female genitalia and renal failure.
The possible etiologies of a DSD are shown on Table 19.1 [19, 20].
Table 19.1
DSD Classification
DSD due disorders gonadal development | |
Ovotestis (karyotype 46XX, 46XY, 46XX/46XY) | |
Gonadal Dysgenesis (karyotypes 45X/46XX, 45X/46XY, etc.) | |
Gonadal Dysgenesis associated with syndromic phenotype (45X, 47XXX, etc.) | |
Embryonic Testicular Regression Syndrome (karyotype 46XY) | |
Others: SF1, WT1 (WAGR, Denys–Drash and Frasier syndromes) | |
46XX DSD | 46XY DSD |
Fetal | Defect of Androgens Synthesis |
Congenital Adrenal Hyperplasia (CAH) 21α-hydroxylase deficiency (CYP21A2) 11β-hydroxylase deficiency (CYPB11B1 e CYPB11B2) 3β-hydroxysteroid dehydrogenase 2 deficiency | Associated with cholesterol synthesis defects (Smith–Lemli–Opitz syndrome) |
STAR deficiency and P450scc deficiency | |
3β-hydroxysteroid dehydrogenase 2 deficiency 17α-hydroxylase and 17,20lyase deficiency (CYP17) | |
P450-oxidoreductase (POR) | |
Maternal Fetal | Defect Testicular Steroidogenesis |
P450-oxidoreductase deficiency (POR) | Isolated 17,20lyase deficiency |
Aromatase deficiency(CYP19) | 17α-hydroxysteroid dehydrogenase 3 deficiency |
Maternal Hyperandrogenism | Defect of Testosterone Metabolism |
Endogenous tumors (Adrenal and ovaries) | 5α-reductase type 2 deficiency |
Exogenous (progestin and others) | Defect of Androgen Action |
CAIS | |
PAIS | |
Persistence of Müllerian Ducts Syndrome | |
Others | |
Endocrine disruptors | |
Hypospadias | |
Cryptorchidism | |
Approach to 46XXDSD
Individuals with 46XX DSD will typically have female internal genitalia (Müllerian structures and ovaries). The differential diagnosis of 46XX DSD includes the disorders of gonadal development previously mentioned. The androgen excess of the fetus is due to CAH or other enzymes or maternal fetal disorders or gestational hyperandrogenism [11, 21].
Androgen Excess of the Fetus
CAH: Is a group of autosomal recessive disorders characterized by enzymatic defect in steroidogenesis (Fig. 19.7). The six forms of CAH are summarized in Table 19.2.
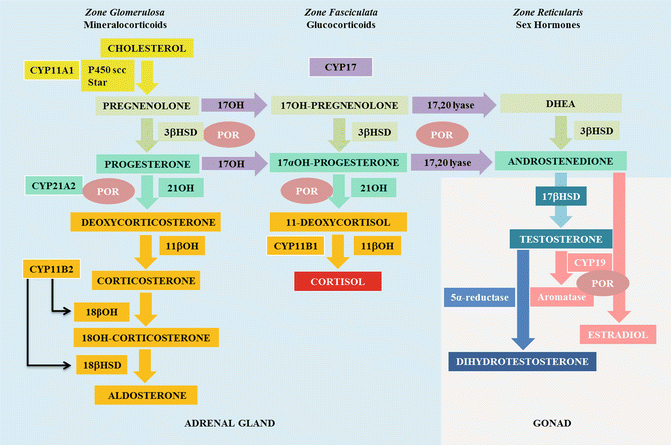
Fig. 19.7
Steroidogenesis. 3β-hydroxysteroiddesydrogenase (3βHSD), 17 hydroxylase (17OH), 18β hydroxylase (18βOH), 18βhydroxydesidrogenase (18βHSD)
Table 19.2
Congenital Adrenal Hyperplasia
Enzymatic defect
Lipoid hyperplasia
3β − hydroxysteroid dehydrogenase 2
21α-hydroxylase
11β-hydroxylase
17α-hydroxylase
P450-oxidoreductase
Gene
StAR CYP11A1
HSD3B2
CYP21A2
CYP11B1
CYP17
POR
Gene localization
8p11.2/15q23-24
1p13.1
6p21.3
8q24.3
10q24.3
7q11.2
Ambiguous genitalia
Male
Male
Female
Female
Male
Male/female
Postnatal virilization
No
Yes
Yes
Yes
No
No
Frequency
Rare
Rare
1:14,000
1:100,000
1:50,000
Rare
Hormones
Glucocorticoids
↓
↓
↓
↓
↓
Normal
↓ after ACTH
Mineralocorticoids
↓
↓
Normal or ↓
↑
↓
Normal or ↓
AndrogensRelated posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree

 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access