Low global arginine bioavailability (GAB) is associated with numerous complications of SCD including early mortality. Mechanisms of arginine dysregulation involve a complex paradigm of excess activity of the arginine-consuming enzyme arginase, elevated levels of asymmetric dimethylarginine, altered intracellular arginine transport, and nitric oxide synthase dysfunction. Restoration of GAB through exogenous supplementation is therefore, a promising therapeutic target. Studies of arginine therapy demonstrate efficacy in treating patients with leg ulcers, pulmonary hypertension risk, and pain. Co-administration with hydroxyurea increases levels of nitrite and fetal hemoglobin. Addressing the alterations in the arginine metabolome may result in new strategies for treatment of SCD.
Key points
- •
Low global arginine bioavailability is associated with severe pain, pulmonary hypertension risk, and early mortality in sickle cell disease (SCD).
- •
Mechanisms of arginine dysregulation in SCD involve a complex paradigm of excess arginase activity, elevated levels of asymmetric dimethylarginine (an arginine analogue and nitric oxide synthase inhibitor), altered intracellular arginine transport, renal dysfunction (which disrupts normal de novo arginine synthesis in the kidneys), and nitric oxide synthase dysfunction.
- •
Arginine therapy shows promise for the treatment of leg ulcers, pulmonary hypertension, and vasoocclusive pain episodes in patients with SCD in preliminary studies.
- •
Parenteral arginine (100 mg/kg per dose 3 times a day) decreased total opioid use by more than 50% and was associated with lower pain scores at discharge in children with SCD hospitalized for pain compared with placebo in a recently published randomized placebo-controlled trial of arginine therapy.
An altered arginine metabolome in sickle cell disease
Normal arginine metabolism is impaired in sickle cell disease (SCD) for a variety of reasons discussed in this review that contribute to endothelial dysfunction, vasoocclusion, pulmonary complications, and early mortality ( Fig. 1 ). The altered arginine metabolome differs in children compared with adult patients. Adults with SCD are arginine deficient at steady state, whereas children tend to have plasma levels that are similar to normal controls. An arginine deficiency develops over time and is influenced by acute events and chronic end-organ damage. Plasma arginine concentration decreases significantly, however, in both adults and children during vasoocclusive pain episodes (VOE) and acute chest syndrome (ACS) and is associated with low plasma nitric oxide (NO) metabolite (NO x ) levels. Low plasma arginine levels predicted the need for hospitalization in children evaluated in an emergency department for pain and normalized with clinical recovery. Although mechanisms of arginine dysregulation are complex and multifactorial, they can be overcome through arginine supplementation, a phenomenon known as the “arginine paradox.” Short-term oral arginine therapy significantly improved estimated pulmonary artery systolic pressures in patients with SCD at risk for pulmonary hypertension in a small pilot study, whereas parenteral arginine significantly decreased total opioid use and improved pain scores in children with SCD hospitalized for VOE compared with placebo in a recently published randomized controlled trial. Case reports also suggest a potential role of arginine for the treatment of leg ulcers. Addressing the alterations in arginine metabolism may result in new strategies for the treatment of SCD.
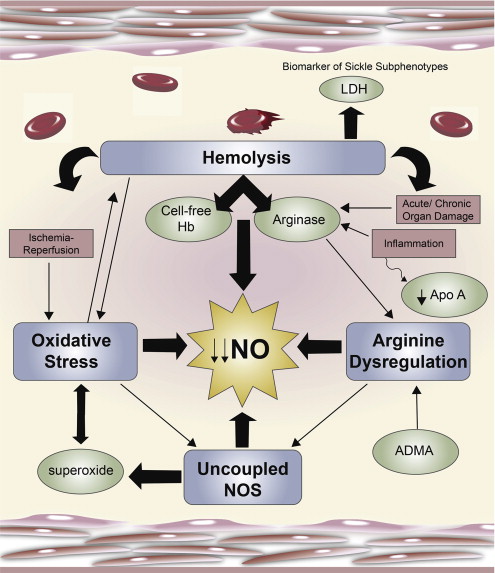
An altered arginine metabolome in sickle cell disease
Normal arginine metabolism is impaired in sickle cell disease (SCD) for a variety of reasons discussed in this review that contribute to endothelial dysfunction, vasoocclusion, pulmonary complications, and early mortality ( Fig. 1 ). The altered arginine metabolome differs in children compared with adult patients. Adults with SCD are arginine deficient at steady state, whereas children tend to have plasma levels that are similar to normal controls. An arginine deficiency develops over time and is influenced by acute events and chronic end-organ damage. Plasma arginine concentration decreases significantly, however, in both adults and children during vasoocclusive pain episodes (VOE) and acute chest syndrome (ACS) and is associated with low plasma nitric oxide (NO) metabolite (NO x ) levels. Low plasma arginine levels predicted the need for hospitalization in children evaluated in an emergency department for pain and normalized with clinical recovery. Although mechanisms of arginine dysregulation are complex and multifactorial, they can be overcome through arginine supplementation, a phenomenon known as the “arginine paradox.” Short-term oral arginine therapy significantly improved estimated pulmonary artery systolic pressures in patients with SCD at risk for pulmonary hypertension in a small pilot study, whereas parenteral arginine significantly decreased total opioid use and improved pain scores in children with SCD hospitalized for VOE compared with placebo in a recently published randomized controlled trial. Case reports also suggest a potential role of arginine for the treatment of leg ulcers. Addressing the alterations in arginine metabolism may result in new strategies for the treatment of SCD.
Altered NO homeostasis
NO has been well described in the literature as an important signaling molecule involved in the regulation of many mammalian physiologic and pathophysiologic processes. As one of the most potent vasodilators known, NO is essential to vascular homeostasis. It plays a critical role in the maintenance of vasomotor tone, limits platelet aggregation and ischemia-reperfusion injury, modulates endothelial proliferation, and has antiinflammatory properties.
NO is produced by a family of NO synthase (NOS) enzymes that metabolize l -arginine through the intermediate N-hydroxy- l -arginine (NOHA) to form NO and l -citrulline using oxygen and nicotinamide adenine dinucleotide phosphate as cosubstrates. NO causes vasodilation through the activation of soluble guanylate cyclase to produce the intracellular messenger cyclic guanylate monophosphate. Increased consumption and decreased production of both NO and arginine contribute to complications associated with SCD.
Hemolysis will significantly compromise NO bioavailability in SCD with the release of cell-free hemoglobin (Hb) that rapidly scavenges NO. Under normal conditions, Hb is safely packaged within the erythrocyte plasma membrane; however, during hemolysis it is decompartmentalized and released into plasma where it rapidly reacts with and destroys NO. This process results in abnormally high NO consumption and the formation of reactive oxygen species, ultimately inhibiting vasodilation. This phenomenon has also been implicated as a mechanism of NO depletion in the red cell storage lesion and other hemolytic conditions, such as paroxysmal nocturnal hemoglobinuria and malaria. The simultaneous release of the arginine-consuming enzyme arginase also found within human erythrocytes will further compromise the obligate substrate for NO production during hemolysis, as arginase is released into circulation in active form, shifting metabolism of l -arginine to l -ornithine and urea.
Altered arginine homeostasis
As the obligate substrate for NOS, l -arginine bioavailability plays a key role in determining NO production and depends on pathways of biosynthesis, cellular uptake, and catabolism by several distinct enzymes ( Fig. 2 ), including those from the NOS and arginase enzyme families. Little is known about arginine metabolism to creatine and agmatine in SCD.
Biosynthesis of the semiessential amino acid occurs in a stepwise fashion though what is called the intestinal-renal axis . l -glutamine is absorbed from the small intestine and converted to l -citrulline by the enterocytes. l -citrulline is also synthesized from l -ornithine by ornithine carbamoyl transferase and carbamoyl phosphate synthetase 1 in hepatocytes as part of the urea cycle, as well as in the intestine. l -arginine is produced from l -citrulline by cytosolic enzymes argininosuccinate synthetase 1 and argininosuccinate lyase in kidneys. When l -arginine is subsequently metabolized to NO via NOS, l -citrulline is again produced and can be used for recycling back to l -arginine, which may be an important source of l -arginine during prolonged NO synthesis by inducible nitric oxide synthase.
Arginine becomes an essential amino acid under conditions involving an increased catabolic state, such as sepsis, burn injury, and trauma, when the capacity of endogenous arginine synthesis is surpassed. It is likely that SCD represents another catabolic state whereby the body’s ability to maintain an arginine balance is disrupted, with several mechanisms involved summarized in Box 1 . In addition to acute pain events, low global arginine bioavailability is also found in adults at steady state with a hemolytic subphenotype that includes risk for leg ulcers, pulmonary hypertension risk, and priapism and is associated with high plasma lactate dehydrogenase levels. Of concern, low arginine bioavailability is associated with early mortality in both adults and children with SCD. Accumulating data also support its role as a biomarker of vasculopathy that goes beyond SCD, associated with malaria-related mortality ; diabetes control ; severe asthma ; pulmonary hypertension risk ; coronary artery disease; and major adverse cardiovascular events, including stroke and mortality in patients screened for cardiovascular disease. Coined the global arginine bioavailability ratio , this biomarker, defined as the ratio of arginine/(ornithine + citrulline), has emerged as a more robust predictor of cardiovascular disease than cholesterol. These findings suggest that adequate arginine bioavailability is critical for survival and provide clinicians with an objective index of disease severity.
- •
Excess arginase concentration and activity
- •
Intracellular arginine transport dysfunction
- •
Renal dysfunction
- •
Exogenous NOS inhibitors (arginine analogues)
Increased Arginase Activity and Concentration
Arginase is an essential enzyme in the urea cycle, responsible for the conversion of arginine to ornithine and urea. The NOS and arginase enzymes can be expressed simultaneously under a wide variety of inflammatory conditions, resulting in competition for their common substrate. Although the affinity (the Michaelis constant K m ) of l -arginine for arginase is in the low micromolar range compared with the low millimolar range for NOS, substrate competition does occur between arginase and NOS because the maximum rate achieved (Vmax) of arginase is 1000-fold higher. Two forms of arginase have been identified: type 1, a cytosolic enzyme highly expressed in the liver, and type 2, a mitochondrial enzyme found predominantly in the kidney, prostate, testis, and small intestine. Arginase-1 is also present in human red blood cells. Plasma arginase activity is elevated in SCD as a consequence of inflammation; liver dysfunction; and, most significantly, by the release of erythrocyte arginase during intravascular hemolysis, which has been demonstrated by the strong correlation between plasma arginase levels and cell-free Hb levels and other markers of increased hemolytic rate. In addition, arginase activity is higher in the erythrocytes of patients with SCD compared with normal controls and strongly correlates to plasma arginase activity. Upregulated expression of arginase-1 also results in increased proliferation rates of vascular smooth muscle and endothelial cells and, in this capacity, may further contribute to vasculopathy in addition to its unique role during hemolysis. When arginine is catalyzed to NO, NOS produces the intermediate product NOHA. NOHA is a potent arginase inhibitor, reflecting complicated feedback mechanisms in place to maintain homeostasis, with both NOS and arginase playing a regulatory role in NO production. Because there is only limited arginase-1 found in the murine erythrocyte compared with human red blood cells, the major sources of increased arginase activity in the sickle cell mouse originate from cells other than the erythrocyte. It is unfortunately not feasible to extrapolate the contribution of erythrocyte arginase release to complications of hemolysis in human SCD from the sickle cell mouse model.
Whether inflammatory or hemolytic in origin, arginase will redirect the metabolism of arginine to ornithine and the formation of polyamines and proline, which are essential for smooth muscle cell growth and collagen synthesis. By creating a shift toward ornithine metabolism, arginase can trigger a process that contributes to the vascular smooth muscle proliferation and airway remodeling that occur in pulmonary hypertension and asthma, common comorbidities in SCD.
Intracellular Arginine Transport
The primary source of l -arginine for most cells is cellular uptake via the Na-independent cationic amino acid transporter (CAT) proteins of the y + -system. l -arginine uptake via the y + -system can be inhibited by other amino acids, such as l -ornithine and l -lysine. Therefore, an arginase-triggered increase in ornithine will further impact arginine transport and bioavailability. Plasma arginine concentration in adults with SCD is approximately 40 to 50 μM at baseline, well less than the K m for CAT (100–150 μM). Even modest fluctuations in extracellular arginine concentration, may significantly impact cellular arginine uptake and bioavailability. Alterations in arginine transport have been demonstrated in several disease states, including septic shock, hypertension, diabetes mellitus, and asthma, although little is known about CAT and arginine transport in SCD.
Renal Dysfunction
Renal dysfunction is common in SCD and will further diminish global arginine bioavailability through the loss of de novo arginine synthesis from citrulline, which occurs primarily in the kidney. Renal dysfunction impairs the major route for endogenous arginine biosynthesis, thereby contributing to a global reduction in arginine bioavailability.
Endogenous NOS Inhibitors
Low arginine bioavailability may be exacerbated further by the presence of elevated asymmetric dimethylarginine (ADMA), an endogenous NOS inhibitor that competes with l -arginine for binding to NOS. Well established as a biomarker of cardiovascular disease and endothelial dysfunction, it may also contribute to inflammation, collagen deposition, and nitrosative stress in SCD. Circulating ADMA levels are elevated in SCD and have been implicated in the pathophysiology of asthma, systemic and pulmonary hypertension, and risk of early mortality. The most elevated ADMA level occurred in patients with SCD with the highest hemolytic rate and was associated with pulmonary hypertension risk reflected by a high tricuspid regurgitant jet velocity (TRV) and mortality. High levels of ADMA can also contribute to NOS uncoupling. Landburg and colleagues recently demonstrated that elevated ADMA levels in patients with SCD did not increase over baseline during VOE. Although they conclude that there is no primary role for ADMA during acute pain, given that arginine bioavailability decreases significantly during VOE and ACS, an increase in the ratio of ADMA to arginine may have some impact on global arginine bioavailability and endothelial dysfunction that should be explored further.
Impact of arginine therapy on NO production: a potential explanation for a varied response to therapy
Mechanistically, oral arginine (100 mg/kg per dose) acutely increases both plasma and exhaled NO when administered to African American healthy control subjects within 2 hours. When arginine is given to patients with SCD at steady state, however, a paradoxic decrease in plasma NO x occurs that is not overcome by higher doses, clearly indicating that arginine is metabolized differently in SCD compared with control subjects. However, when arginine is given during VOE, a condition associated with an acute arginine deficiency, a robust dose-dependent increase in NO x occurs.
These early observations may account for the negative outcome of the Comprehensive Sickle Cell Centers’ (CSCC) arginine trial that targeted patients with SCD at steady state, particularly because the primary outcome measure of that study was an increase in plasma NO x , despite preliminary data showing a paradoxic decrease in NO x when oral arginine is given to patients with SCD at steady state. Also, low-dose arginine therapy is likely to be subtherapeutic in SCD and may represent an additional flaw in the CSCC prophylactic arginine trial design because doses used were close to placebo based on the cardiovascular literature. Previous studies have shown that low-dose arginine is unlikely to impact NO synthesis, an observation confirmed in the CSCC study.
Based on preliminary pharmacokinetic studies, peak plasma arginine concentration after oral arginine (100 mg/kg) is significantly higher during SCD steady state compared with VOE, although levels are similar by 4 hours. Healthy controls reach a peak arginine level between 1 to 2 hours that is maintained at 4 hours and does not trend down as in SCD. Accelerated arginine metabolism or consumption occurs during VOE compared with steady state despite the same oral arginine dose given. The author also found that the capacity of arginine to increase NO x production in patients with SCD with pain is dose dependent. Higher concentrations of plasma arginine are likely needed to overcome multifactorial effects, including the impact of arginase and ADMA on global arginine bioavailability. Therefore, low doses are unlikely to achieve a maximal benefit. However, the long-term safety of doses greater than 100 mg/kg per dose 3 times per day is unknown in SCD, even though a 1-time dose of 30 g is commonly used for growth hormone stimulation testing with an excellent safety profile. Because the arginine formula generally available is l -arginine hydrochloride, the wisdom of higher doses is questionable given the potential to induce acidosis with repeated dosing over time and must be taken into consideration. However, a single loading dose of 200 to 300 mg/kg may provide an additional benefit during VOE given the pharmacokinetics data showing increased arginine consumption in patients with pain compared with steady state and warrants further study. However, to date, the safety of 100 mg/kg per dose 3 times a day (300 mg/kg/d total) has been well established in the literature and has been found efficacious in the author’s experiences with SCD.
These data highlight the importance of careful identification of ideal outcome measures, optimal study drug dosing, and enrollment of targeted clinical phenotypes to insure comparison of apples to apples in light of growing evidence of metabolic variations among patients with SCD that may manifest with a specific clinical profile.
In transgenic mouse models of SCD, l -arginine supplementation inhibits the red cell Gardos channels ; reduces red cell density ; improves perfusion; and reduces lung injury, microvascular vasoocclusion, and mortality. Arginine also increases erythrocyte glutathione levels in both mouse and human trials and may downregulate inflammatory pathways. In addition, arginine is a key substrate in creatine synthesis, an important metabolic pathway not yet sufficiently studied in SCD that may be impacted by an arginine-deficient state. Although the role of NO in SCD has become controversial, these studies demonstrate that the mechanistic impact of arginine goes beyond NO.
Arginine coadministration with hydroxyurea
Coadministration of oral arginine with hydroxyurea (HU) ameliorated the paradoxic decrease in plasma NO x observed in patients with SCD at steady state compared with arginine monotherapy. A recently published study performed in Brazil adds to the growing body of literature in support of arginine coadministration with HU. Twenty-one adult patients with SCD were randomized to receive HU alone (500–1500 mg/d; n = 9) or HU + arginine (250 mg/d; n = 12) for 12 weeks. An increase in levels of nitrite and fetal Hb was observed in the arginine/HU arm compared with patients receiving HU alone, despite the low dose of arginine used. Arginine therapy together with HU may be superior to either single intervention.
Arginine therapy for clinical complications of SCD
Leg Ulcers
Chronic refractory leg ulcers are a debilitating and painful complication of SCD. To date, there is no specific Food and Drug Administration (FDA)–approved therapy for leg ulcers, and most patients undergo multiple treatments of surgical debridement and grafting and courses of topical and systemic treatments with only anecdotal evidence of improvement. Rapid healing of leg ulcers was initially reported with parenteral arginine butyrate in both SCD and thalassemia. Although improvement of leg ulcers occurred in patients despite little change in fetal hemoglobin, considered to be butyrate’s mechanism of action, it is not possible to determine from these observations whether the benefits were in fact from the butyrate or arginine component of treatment. Rapid healing of chronic recalcitrant leg ulcers was also observed after 5 days of oral l -arginine-hydrochloride (100 mg/kg 3 times a day) in a pilot study treating patients with SCD with pulmonary hypertension risk, suggesting a therapeutic potential for the arginine component. A randomized-controlled phase II trial of parenteral arginine butyrate confirmed the initial anecdotal observations. Eligible patients included adults with SCD suffering from extremity ulcers refractory to standard care for at least 6 months. Many of the enrolled patients had suffered from refractory ulcers for many years and had tried multiple therapies. Twenty-three patients with 25 refractory leg ulcers present were randomized to receive standard local care alone (the control arm, n = 12) or standard care together with parenteral arginine butyrate administered 5 days per week for 12 weeks (n = 11, patients with 37 leg ulcers). A higher percentage of ulcer proportions healed after 3 months in the treatment arm compared with the control arm (78% vs 24%, P <.001). Additional case reports of oral arginine therapy anecdotally improving recalcitrant leg ulcers in SCD support the need for further study.
Pulmonary Hypertension Risk
The cause of pulmonary hypertension is multifactorial; however, there is growing evidence that the disease process, in part, involves altered arginine metabolism or decreased bioavailability. Several human studies have demonstrated therapeutic benefits of arginine therapy for both idiopathic and secondary pulmonary hypertension, although other studies have demonstrated little effect. A single arginine infusion (500 mg/kg over 30 minutes) decreased pulmonary vascular resistance and improved blood oxygenation in infants with persistent pulmonary hypertension of the newborn. Mehta and colleagues found that an infusion of arginine (500 mg/kg over 30 minutes) reduced mean pulmonary artery pressures by nearly 16% and pulmonary vascular resistance by 27.6% in 10 patients with pulmonary hypertension of various origins. Nagaya and colleagues demonstrated that oral arginine (50 mg/kg) produced a 9% decrease in mean pulmonary artery pressure and a 16% decrease in pulmonary vascular resistance 60 minutes after supplementation in 10 patients with pulmonary hypertension of mixed cause compared with 9 patients receiving placebo. One-week continued supplementation of arginine (150 mg/kg 3 times a day) resulted in a significant improvement in cardiopulmonary exercise testing; however, the impact on hemodynamics was not reassessed. In contrast, an acute arginine infusion (12.6 g over 90 minutes) in 4 patients with idiopathic pulmonary artery hypertension demonstrated little impact on pulmonary hemodynamics and a significant decrease in systemic resistance in 2 of the patients, whereas an arginine infusion (500 mg/kg over 30 minutes) given to 5 controls and 5 patients with systemic sclerosis and pulmonary artery hypertension demonstrated no significant effect on systemic or pulmonary hemodynamics in either group.
Pulmonary hypertension is a common complication in hemoglobinopathies. A similar pattern of altered arginine metabolism is seen in both SCD and thalassemia. The prevalence of pulmonary hypertension in SCD has generated a great deal of controversy ; however, recent screening studies in SCD that include right heart catheterization (RHC) for patients at an increased risk of pulmonary hypertension based on an elevated TRV on Doppler echocardiography demonstrate that 6% to 11% of adults with SCD have pulmonary hypertension defined by a mean pulmonary artery pressure greater than or equal to 25 mm Hg. This prevalence of 6% to 11% pulmonary hypertension in SCD is much higher than the prevalence of 0.0015% in the general population, 0.5% in patients with human immunodeficiency virus, and 7.8% in patients with scleroderma. Given the global burden of SCD affecting millions of patients, SCD is one of the most common causes of pulmonary hypertension worldwide. In light of the high mortality risk associated with pulmonary hypertension in SCD, novel therapies are needed, although studies to date with traditional FDA-approved pulmonary artery hypertension medications, including bosentan and sildenafil, have been disappointing. In order to provide some recommendations for clinicians, consensus-based guidelines for the diagnosis and treatment of pulmonary hypertension of SCD have recently been created.
Similar to the original observations with respect to leg ulcers, anecdotal improvement in estimated pulmonary artery pressure was observed after treatment with arginine butyrate in patients with SCD and pulmonary hypertension risk who were on HU therapy. In a later study of 10 patients with SCD and high risk for pulmonary hypertension determined by an elevated TRV, oral l -arginine hydrochloride supplementation (100 mg/kg 3 times a day) produced a 15.2% mean reduction in pulmonary artery pressures estimated by Doppler echocardiography and improved venous oxygen saturation measured by co-oximetry after 5 days of treatment ( Fig. 3 ). Plasma arginine concentration more than doubled in all compliant patients. Only one patient did not demonstrate a change in pulmonary artery systolic pressure after supplemental arginine; however, noncompliance was indicated by a low plasma arginine concentration at the end of the study in addition to the patient’s admission to not taking the study drug. Arginase activity was also found to be high in this cohort of patients, a phenomenon observed in pulmonary hypertension of various causes.
Related posts:
 Ischemia-reperfusion Injury in Sickle Cell Anemia
Ischemia-reperfusion Injury in Sickle Cell Anemia
 Gene Therapy for Hemoglobinopathies
Cellular Adhesion and the Endothelium
Hemoglobin S Polymerization and Red Cell Membrane Changes
Gene Therapy for Hemoglobinopathies
Cellular Adhesion and the Endothelium
Hemoglobin S Polymerization and Red Cell Membrane Changes
 Inflammatory Mediators of Endothelial Injury in Sickle Cell Disease
The Role of Adenosine Signaling in Sickle Cell Therapeutics
Inflammatory Mediators of Endothelial Injury in Sickle Cell Disease
The Role of Adenosine Signaling in Sickle Cell Therapeutics
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


