17.1 Introduction
The importance of maintaining good nutrition during pregnancy is well established, as reflected in the increased dietary recommendations for many micronutrients which are considered necessary to cover the extra nutrient demands of pregnancy and lactation. However, since the 1980s there has been more recognition of the importance of ensuring a good nutritional status in the mother, not only during pregnancy and lactation, but also before conception and in the very early weeks of pregnancy. In particular, maternal folate status is now known to have a major impact on early development of the embryo up to the first 4 weeks of pregnancy, a time when many women will not be aware that they are pregnant. There is now conclusive evidence with respect to the benefits at this time of optimal maternal folate status in the prevention of neural tube defects (NTD). Although this knowledge is universally accepted across the scientific and medical communities, it is less clear how best the official recommendations can be translated into public health policy to optimize folate status in women who may become pregnant. What is evident is that the diet currently consumed by most women of childbearing age in European countries fails to provide levels of intake of folate and the metabolically related B-vitamins that are associated with optimal maternal folate status in early pregnancy and therefore the lowest risk of NTD.
As pregnancy progresses, folate continues to play an important role, but after the early weeks the emphasis is focused on protecting the mother from developing folate deficiency of pregnancy. Maternal folate deficiency not only has consequences for the mother’s own health, but has also been shown to result in fetal growth retardation, low birth weight (LBW) and neonatal folate deficiency, with important implications for the health of the neonate and infant. Thus, throughout pregnancy, folate plays key roles in terms of maternal, fetal and neonatal health. The public health challenge now facing policy makers and others is how best to achieve optimal folate status in women of reproductive age.
This chapter will focus on the role of folate and metabolically related B-vitamins during preconception and pregnancy in protecting against adverse outcomes for both the mother and the baby.
17.2 Biochemical basis for the role of folate in adverse outcomes of pregnancy
As discussed above, there is conclusive evidence that any reduction in folate status has an associated increased risk of an NTD and possibly also other birth defects. How this reduced status increases risk, and its correction by extra folic acid/folate reduces risk, is not understood. However, to avail of this potentially substantial public health benefit in an optimal way it is important to increase our understanding of the event or events involved and how they are modulated by folate and related nutrients. To make this possible, a comprehensive understanding of the biochemistry of this area, which is largely understood, is an essential starting point.
Metabolic role of folate cofactors and metabolically related B-vitamins
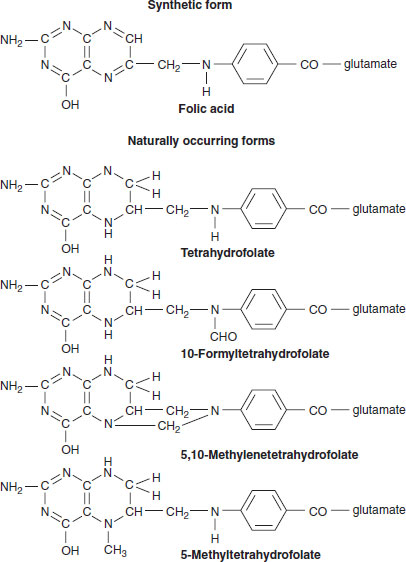
The folate cofactors exist in cells as their metabolically active tetrahydro forms (Figure 17.1). Their metabolic role is as cofactors to the several folate-dependent enzymes found in all cells (Figure 17.2). This role consists principally of the metabolic transfer or donation of carbon-1 units. As such, they are involved in two cycles in cells, the DNA biosynthesis cycle and the methylation cycle. The source of these carbon-1 units is directly from the carbon-3 of serine via serine hydroxymethyltransferase, leading to the synthesis of 5,10-methylenetetrahydrofolate. A more significant source of carbon-1 units is from serine, but after its uptake and metabolism by mitochondria. The result is that formate is produced in the mitochondria from cytoplasmic serine. This formate emerges from the mitochondria and is used by the trifunctional enzyme to make folates with carbon-1 units, namely 10-formyltetrahydrofolate and 5,10-methylenetetrahydrofolate. Thus, there are two possible ways of making these carbon-1 folate cofactors in cells. These go on to be the carbon-1 donors in purine and pyrimidine biosynthesis, respectively, as discussed below (Figure 17.2). They can also be converted to 5-methyltetrahydrofolate.
The DNA biosynthesis cycle
The biosynthesis of purine involves two enzymes that are folate dependent. The folate cofactor 10-formyltetrahydrofolate is involved in the synthesis of purines and the folate cofactor 5,10-methylenetetrahydrofolate is involved in pyrimidine synthesis (Figure 17.2). It is important to recognize that the folate cofactor conversions are part of these two cycles with the continuous regeneration of tetrahydrofolate, which can then accept a further carbon-1 group, as described above. Folates are present in only tiny amounts in cells and the different forms are continuously recycled. Anything that interrupts their recycling, be it a deficiency of folate, a major inborn error of metabolism or a less significant change in metabolism due to a polymorphism in any enzyme involved in these or other cycles, will quickly reduce or even stop folate-related biosynthesis and affect purine and pyrimidine biosynthesis. This, in turn, will diminish DNA biosynthesis and with it cell division. While this will affect all replicating cells it will be most apparent in those cells with rapid replication rates such as the erythroid series making red cells, with a reduction in their rate of cell division progressing to a macrocytic anemia. This anemia is further characterized by the arrest of red cell maturation in the bone marrow. These abnormal red cell precursors, called megaloblasts, only arise when the rate of purine and pyrimidine biosynthesis is decreased so as to compromise the normal rate of mitotic cell division. This, in turn, results in an abnormally large, poorly differentiated nucleus, which is characteristic of a megaloblast. These megaloblasts are unique to impaired purine and pyrimidine biosynthesis. They only arise directly through folate deficiency or indirectly through interrupted folate metabolism which occurs consequent to vitamin B12 deficiency. The only other cause of megaloblastic anemia is the use of drugs which either directly (cytosine arabinoside) or indirectly (methotrexate) inhibit DNA biosynthesis. Thus, folates are essential for DNA biosynthesis and cell replication and could be said to participate in the DNA cycle.
Figure 17.1 Synthetic and naturally occurring forms of the folates.
The methylation cycle
The other role of the folates is less obvious but no less important. All cells contain a range of enzymes called methyltransferases. As this name suggests, their function is to transfer a methyl (—CH3) group. The range of substrates for these methylations is very wide indeed. They include:
- methylation of the hormone dopa to methyl-dopa, as one of its methods of inactivation
- methylation of the phospholipid phosphotidylethanolamine via methyltransferase
- methylation of cytosine residues of DNA, which has the important functional effect of preventing the replication of such methylated DNA
- methylation of proteins, changing their stability (e.g. myelin basic proteins) or their function (isoprenoid methyltransferase changes the function of a G-protein involved in cell signaling).
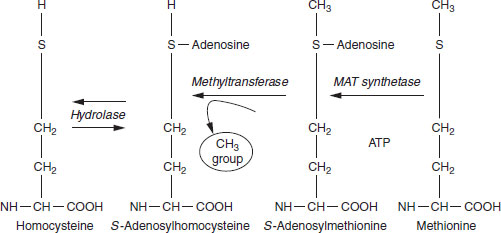
All methyltransferases must have the methyl group presented to their active site in a high-energy activated form. This is the methionine derivative S-adenosylmethionine (abbreviated to SAM or AdoMet). It and its related structures are shown in Figure 17.3. A product of the methyltransferase reactions is SAM minus its methyl group, namely S-adenosylhomocysteine (SAH or AdoHcy). This product, SAH, is subject to only one fate: it is rapidly hydrolyzed to homocysteine and adenosine, the kinetics of which result in the level of SAH being kept extremely low in all cells except in abnormal circumstances. This is important functionally because SAH is a potent inhibitor of all methyltransferases, with one exception. Different methyltransferases may be switched off at different levels of accumulation of SAH, depending on their functional importance. If for any reason, such as during deficiency of folate, vitamin B6 or vitamin B12, homocysteine accumulates in the cell, it will inevitably lead to an elevation of intracellular SAH with a consequence of inhibition of several methyltransferases. Depending on the methyltransferase that is being inhibited, this could have a range of consequences discussed below under clinical and subclinical signs of folate or vitamin B12 deficiency.
Figure 17.2 Metabolism of the folate cofactors and their role in DNA biosynthesis and in the methylation cycle.
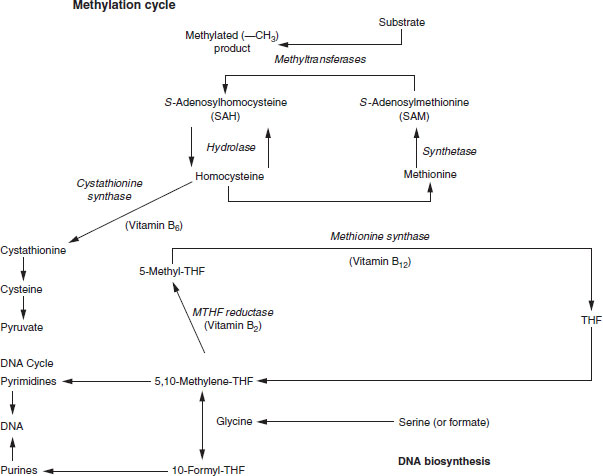
Figure 17.3 Chemical conversions involved in the methylation cycle resulting in the utilization of a methyl (—CH3) group by the cellular methyltransferase.
The link between the DNA and methylation cycles
To maintain the activity of the methyltransferase, each cell would have two options. It could continue to take up methionine from the plasma, activate it to SAM and use its methyl group for a methylation reaction. This would leave it with the burden of generating an SAH and a homocysteine every time this happened, with the necessity of either degrading or perhaps wastefully exporting this homocysteine back into the plasma. The alternative, which is used by most cells, is to remethylate the homocysteine back to methionine, thus completing the cycle. Cells carry out this methylation of homocysteine back to methionine using the vitamin B12-dependent enzyme methionine synthase. The immediate source of this methyl group is 5-methyltetrahydrofolate. This cosubstrate with homocysteine must be supplied on a continuous basis in all cells. Thus, the methylation cycle continuously uses or consumes methyl groups by donating them to the dozens of methyltransferases in the cell. The input of these methyl groups requires that they are presented attached to the folate cofactor as 5-methyltetrahydrofolate. The origin of these methyl groups is an enzyme, 5,10-methylenetetrahydrofolate reductase (MTHFR), which links the DNA cycle with the methylation cycle. This key enzyme can direct carbon-1 groups away from DNA biosynthesis and supply them instead to the methylation cycle. The carbon-1 that is pulled off is the folate cofactor, 5,10-methylenetetrahydrofolate, which is directly involved in the biosynthesis of DNA. The enzyme MTHFR reduces 5,10-methylenetetrahydrofolate to 5-methyltetrahydrofolate. This enzyme is thus a key link between these two important cycles. It is highly controlled. Its activity in all cells except for those of the liver is controlled by intracellular level of SAM. As the intracellular level of SAM begins to drop there is a consequent increase in the activity of the enzyme. The resultant increased level of 5,10-methyltetrahydrofolate facilitates the methylation of homocysteine to methionine by methionine synthase. Once the methionine and consequently the SAM level has been restored in the cell, its rising level progressively inhibits MTHFR.
This method of control and the fact that MTHFR is irreversible in vivo have profound consequences if the activity of methionine synthase is compromised owing, for example, to vitamin B12 deficiency. In that circumstance the interruption of the methylation cycle at the point of this enzyme inevitably leads to the cell running low in SAM to supply its methyltransferases. The proper response is that the resultant reduction in the inhibition of MTHFR will cause diversion of carbon-1 units from the DNA cycle into the methylation cycle by increasing levels of 5-methyltetrahydrofolate as a result of increased activation of MTHFR. However, the only enzyme capable of utilizing this cofactor is methionine synthase, the activity of which is decreased or absent because it requires vitamin B12. Thus, as suggested by the methyl trap hypothesis, 5-methyltetrafolate becomes metabolically trapped in this form. Surprisingly, the cell seems to have no metabolic response or only a wrong metabolic response to such vitamin B12 deficiency. Instead of recognizing that no amount of 5-methyltetrahydrofolate can restart the methylation cycle in the absence of vitamin B12 and methionine synthase, the cell continues to respond to the ever-decreasing SAM level by further increasing the activity of MTHFR. The resultant continuing drawing of the folate cofactors away from the forms found in the DNA cycle into the methodically trapped 5-methyltetrahydrofolate form quickly depletes the former pools of 5,10-methylenetetrahydrofolate and 10-formyltetrahydrofolate and reduces the cells’ capacity to synthesize pyrimidines and purines, and thus DNA. The cell thus has its folate in a form that cannot be used and undergoes a sort of pseudo-folate deficiency, where there is folate in the cell but it cannot be used for DNA biosynthesis or cell replication. The consequence in such circumstances, such as in patients with the vitamin B12 deficiency condition pernicious anemia, is that a macrocytic megaloblastic anemia develops which is indistinguishable from that which occurs in folate deficiency. In addition, even though the level of 5-methlytetrahydrofolate is high in such vitamins B12-deficient cells, it cannot be used to maintain the methylation cycle. This is because its use has an absolute requirement for a functioning methionine synthase. Thus, there is also a progressive reduction in the level of SAM and a reduction in the activity of all of the methyltransferases in the cell. This is probably further exacerbated by the fact that the level of SAM also controls the activity of the enzyme cystathionine synthase, which initiates the catabolism of homocysteine. Low SAM levels effectively shut down this enzyme, decreasing homocysteine catabolism. This accumulated homocysteine cannot be remethylated to methionine. As already mentioned, the equilibrium of SAH hydrolase will favor this homocysteine being converted to SAH. An elevation of SAH would have the additional effect of inhibiting all of the methyltransferase in the cell. Thus, methylation reactions will be compromised in two ways, by a lack of SAM and by inhibition by SAH.
Clinical and subclinical signs of folate or vitamin B12 deficiency
As discussed above, when there is insufficient folate in the diet the level of the folate cofactors necessary to maintain the DNA cycle is insufficient. This, in turn, compromises DNA biosynthesis, causing an arrest in cell division. This is most easily seen in cells that are replicating rapidly, not only because they have a high demand for de novo purine and pyrimidine biosynthesis to maintain DNA biosynthesis, but also because as such cells divide they halve their folate content. The two resultant daughter cells need to reestablish the level of folate to an optimal amount if these new cells are to continue to make DNA at an unhindered level. This is the case for all rapidly dividing cells, such as those of the bone marrow, red cells, granulocytes, lymphocytes and megakaryocytes. One would thus anticipate that folate deficiency, if severe enough and prolonged enough, would lead, respectively, to anemia, infection, a reduced immune response and decreased clotting as a result of a reduction in platelets. Other rapidly dividing cells such as those lining the gut will also be affected, causing malabsorption if the deficiency is severe and prolonged.
Cells that divide are affected, and the degree to which this will happen will depend on their rate of cell division. For example, hepatocytes turn over slowly and thus do not have the joint requirements to make lots of purines or pyrimidines and also to keep reestablishing new folate levels as a result of cell division. In contrast, during embryonic and fetal development and neonatal development, the demand for purines and pyrimidines and the need to take up new folates will be extremely high. Thus, developing structures would be particularly susceptible to reduced folate status. They would also be vulnerable to anything that interfered with the recycling of the folate cofactor, such as reduced vitamin B12 status or a polymorphism that affected a folate-dependent enzyme such as exists for C→T677 MTHFR.
The effects of reduction in the DNA cycle are predictable: decreased purine and pyrimidine biosynthesis followed by reduced DNA biosynthesis and an impaired rate of cell replication most easily seen in the development of characteristic megaloblastic anemia. The consequence of reduction in the methylation cycle, as would be expected also to occur in both folate deficiency and vitamin B12 deficiency, are much more difficult to predict. All methyltransferases in the cell will be compromised.
Thus, there are well-recognized circumstances in which either the DNA cycle or the methylation cycle is compromised. In folate deficiency one would expect both to be compromised and this is most clearly seen in the arrest of cell division causing megaloblastic anemia. One would also expect the methylation cycle to be compromised, and consequently one would expect folate deficiency to lead to the same type of demyelination seen in vitamin B12 deficiency. However, peripheral neuropathy, ataxia and subacute combined degeneration, well-established consequences of vitamin B12 deficiency, are not usually seen in folate deficiency. The reason for this is unclear. A possible explanation is that the central nervous system concentrates folate by a specific transporter, resulting in a higher level in cerebrospinal fluid than exists in the circulating plasma. Perhaps this ensures that nerve tissue continues to have a sufficiency of folate even though there are overt signs and symptoms of deficiency in other tissues and organs. It may be the case that in practice the level of folate usually does not fall to sufficiently low levels in neural tissue to produce a reduction in the methylation cycle sufficient to cause demethylation. In this context, one study found the classical signs associated with vitamin B12 deficiency in subjects who were apparently vitamin B12 replete, but who had chronically extremely low folate levels.
A high level of interest in MTHFR is being generated because a polymorphism for it exists where there is a base change at base pair 677 from a thymidine to a cytosine. This, in turn, changes an amino acid at residue 244 from a glycine to an aspartate. This results in a structural alteration in the resultant enzyme, whereby it loses its cofactor flavin adenine dinucleotide (FAD) from its active site, resulting in instability and an irreversible loss of activity of the enzyme. The resultant phenotype is that those homozygous for the C→T677 variant of MTHFR have significantly reduced levels of the activity of the enzyme in vivo, resulting in a significant elevation of plasma homocysteine. This is greatest in those homozygous for the variant, but also elevated in those heterozygous for the variant. There is a further difference in phenotype with respect to both plasma and red cell folate, with an increasing gradient between those homozygous, heterozygous and wild-type for the variant. The expression of the elevation in plasma homocysteine in those homozygous for the C→T677 variant of MTHFR is very dependent not only on folate status, but also on riboflavin (vitamin B2) status. It has been shown that only those who have the variant accompanied by a reduced riboflavin status show an elevation in plasma homocysteine. It would appear that a sufficiently good supply of riboflavin in the diet produces an amount of its cofactor derivative FAD in the cell that is sufficient to keep the active site of MTHFR with the cofactor bound to it. This, in turn, protects the enzyme from the inactivation that it would experience were the active site to lose its FAD group.
The clinical consequences of this variant are also of great interest. Numerous studies have examined whether it increases the risk of cardiovascular disease (CVD) as would be predicted because of its concurrent elevation in plasma homocysteine, which a body of evidence suggests is associated with CVD. While the results of these studies were mixed, two metaanalyses concluded that the variant does indeed increase CVD risk. As discussed below, it has also been shown conclusively to increase the risk of spina bifida and probably all NTD.
How might reduced folate status increase the risk of neural tube defects?
As discussed above, the metabolic role of the folate cofactors can be divided into two main areas: the DNA cycle (and cell division) and the methylation cycle (and methylation).
Neural tube defects and DNA cycle reduction
The reduced synthesis of DNA associated with reduced folate status or with genetic variants that affect this cycle would be expected to prevent cells dividing at the correct rate. It should be borne in mind that all dividing cells are particularly vulnerable to reduced folate status, not only because of the essential role of folate in making DNA, but also because as cells grow and divide they halve their folate content on each occasion. Thus, for the two daughter cells to work properly they must take up folate from the plasma or their supporting milieu to reinstate the correct level of folate before a further cell division reduces the intracellular folate concentration yet again. If, because of a poor folate supply or an impaired enzyme, this uptake did not keep pace with cell division, such replicating cells would have an increasingly inadequate level of folate and thus be metabolically compromised. This would affect the rate of cell division. Cell division during the early stages of the development of the embryo is extremely rapid. A series of structures is growing in a synchronized manner to bring about the various phases of development. Specifically, the neural plate undergoes a transformation beginning on day 21 postconception and finishing on day 27, with complete closure to form the neural tube. At the same time a protrusion is sent out from this developing structure to form what ultimately becomes the cranium.
An inadequate rate of cell division brought about by an inadequate amount of the necessary folate cofactors could compromise this normal development, resulting in an imperfect closure of the neural tube, causing spina bifida, or a lack of completion of the cranium, causing anencephaly. Similarly, in a pregnancy with inadequate vitamin B12 status some metabolic trapping of folate as 5-methyltetrahydrofolate may occur. The resultant reduction in the forms of folate that participate in purine and pyrimidine, 10-formyltetrahydrofolate and 5,10-methyltetrahydrofolate, respectively, may produce an impaired rate of cell division and a resultant embryo with spina bifida or anencephaly. The effect of increased folate (be it dietary folate or folic acid supplied in supplements and fortified food) would produce higher levels of intracellular folates, thus facilitating DNA biosynthesis and cell division. Similarly, if it is confirmed that vitamin B12 status is a maternal risk factor for NTD, improving intracellular vitamin B12 levels would also ensure that trapping of 5-methyltetrahydrofolate was not an issue.
Because this discussion is about dividing cells, in some instances, very rapidly dividing cells, it must be reemphasized that after each cell division there is a need quickly to reestablish the intracellular folate level halved by this division to its original level. Cells with adequate circulating levels of folate and/or vitamin B12 would be in a position to do this in an optimal way. Similarly, in embryos homozygous for the C→T677 variant of MTHFR adequate riboflavin status would be desirable not only in the cell but also in the circulation, again to replenish the intracellular level after cell division. This polymorphism has been established as contributing a percentage of the population attributable risk for NTD in the order of 13%. A further polymorphism, in the trifunctional enzyme MTHFD-1, has been shown to be a maternal risk factor for an NTD-affected birth, contributing 9% of the population attributable risk. As discussed earlier, the anticipated reduction in the activity of the enzyme would reduce the flow of carbon-1 units from the formate exiting the mitochondria on their way to being converted to 10-formyltetrahydrofolate (for purine biosynthesis) and 5,10-methylenetetrahydrofolate (for pyrimidine biosynthesis). This latter cofactor is also known to be channeled up to the methylation cycle, since radioactive-labeled formate is formed into methionine and methylated cellular derivative in cells in tissue culture. Thus, the polymorphism in MTHFD-1 may also affect methylation. This polymorphism has been shown to be a maternal risk, but there is no increased risk if the embryo is homozygous for this variant. One can only speculate as to how compromising this enzyme in the mother may affect the closing neural tube in the embryo: the mother may supply the embryo with a structure or a metabolite involved in neural tube closure, or she may metabolize an embryonic or a fetal product.
Neural tube defects and methylation cycle reduction
As discussed earlier, the methylation cycle supplies SAM to dozens of methyltransferases. The resultant methylations have an equal number of metabolic effects. Reduced folate status would directly (and reduced vitamin B12 status would indirectly) potentially compromise a whole range of cellular processes. The two risk polymorphisms identified to date, namely C→T677 and MTHFD-1, could also compromise the supply of methyl groups to the methylation cycle.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


