Adult (>16 years of age) soft tissue sarcomas (STS) are a rare, heterogeneous group of nonosseous tumors derived from embryonic mesoderm. It is estimated that there will be 10,600 new cases and 3,820 deaths from STS in the United States in 2010 (1). STS account for about 1% of adult human cancers and 15% of pediatric malignancies, yet distinct prognostic differences between pediatric and adult STS exist. For example, Qaddoumi et al. used the Surveillance, Epidemiology, and End Results program (SEER) from 1973 to 2005 to analyze 2,600 patients, 1,071 adults and 1,529 children with rhabdomyosarcoma. Adults had worse overall survival at 5 years than children (27% vs. 61%) with tumor histology more likely to be the unfavorable pleomorphic subtype. For patients with localized disease, 5-year survival was 82% for the pediatric patients but only 47% for the adults (2). The rate of progression in STS and likelihood of metastases, usually by hematogenous spread to the lungs, is determined primarily by tumor grade. An extent of disease evaluation using chest x-ray and MRI/CT is conducted, and, if no metastatic disease is encountered, wide surgical resection with an effort to achieve negative margins is the mainstay of therapy. Neoadjuvant radiation therapy and/or chemotherapy can be utilized to reduce the local tumor burden in initially unresectable cases with the aim to allow a later complete gross resection. Important prognostic factors include tumor size, grade, depth, site, and histological subtype. Ninety percent of local and systemic recurrences occur within the first 5 years from diagnosis, and 5-year survival rates range between 60% and 80% (3).
Despite their rarity, STS are of great biologic and clinical interest because of recent insight into the molecular and genetic defects that have been defined as well as the many challenges they pose in diagnosis and management. Although these tumors may develop in any anatomic site, 43% occur in the extremities with two thirds of extremity lesions occurring in the lower limb, followed in order of frequency by visceral (19%), retroperitoneal (15%), trunk/thoracic region (10%), and other (13%) (4). STS arising from the genitourinary system (prostate/seminal vesical, bladder, kidney, paratesticular) are exceedingly rare accounting for only 2% of STS and 1% to 2% of malignant GU tumors (5,6). The exact organ or site of origin can sometimes be difficult to determine due to involvement of more than one site or area (i.e., retroperitoneal or kidney primary, base of bladder, prostate, and seminal vesical primary). Although the aforementioned GU organs can be involved and provide a clinical challenge to the urological oncologist and their colleagues, basic principles of management that apply to STS in general also apply to the management of the GU STS.
The world’s literature is replete in case reports and small case series of GU STS, and, instead of attempting to catalogue and thoroughly summarize this literature, we choose to review the basic principles of pathology, molecular biology, and multidisciplinary management of STS in general and discuss the unique factors related to the GU STS. In addition, a comprehensive description of clinical outcomes from the largest reported series of GU STS from our group at Memorial Sloan Kettering Cancer Center (MSKCC) is presented.
PREDISPOSING FACTORS AND MOLECULAR GENETICS
In most STS patients, no specific etiologic factor is found. Multiple predisposing factors have been identified (Table 52.1) including genetic syndromes such as neurofibromatosis, familial adenomatous polyposis, and the Li-Fraumeni syndrome (7,8). Ionizing radiation and lymph edema are well established but uncommon antecedents to the development of soft tissue sarcoma (4). The association with trauma is uncertain as a true causal factor. Chemical carcinogens have also been widely implicated, but the data to support their association are not well founded (9).
Radiation-associated sarcoma (RAS) is of particular concern to oncologists in general and urologists in particular. MSKCC investigators identified 123 of 4,884 (2.5%) patients who developed a sarcoma in a previously radiated field at a median of 103 months (6-534 months) following the completion of radiation therapy delivered most commonly for breast cancer (29%), lymphoma (16%), and prostate cancer (15%). Malignant fibrous histiocytoma (MFH, 23%), fibrosarcoma (15%), and angiosarcoma (15%) were the most common histological subtypes in this group of RAS. The 5-year actuarial survival for the 114 patients who underwent a curative resection was 41%. Factors predictive of a poor prognosis were high-grade tumors, age >60, and a grossly positive resection margin (10). An additional 46 patients with RAS were reported from Belgium with a median time from irradiation to sarcoma of 15 years (range 1-54 years) and overall 5-year survival of 27% following surgical resection (11). Angiosarcomas, a relatively uncommon STS, and atypical vascular lesions (felt to be benign in nature) have been closely associated with breast cancer irradiation sometimes within three years of prior treatment (12). Neuhaus et al. from London reported a series of 67 patients with a median time from radiation to sarcoma of 11 years, a median tumor size of 7 cm, and 56% of tumors were classified as high grade. For patients receiving breast irradiation, angiosarcoma of the chest wall was most common. Despite aggressive surgical resection in this series, the local recurrence (LR) rate was 65% and a 5-year survival was only 45% (13).
TABLE 52.1 PREDISPOSING FACTORS FOR SARCOMAS
Genetic predisposition
Neurofibromatosis (von Recklinghausen disease)
Li-Fraumeni syndrome
Retinoblastoma
Gardner syndrome (familial adenomatous polyposis)
Radiation exposure
Ortho- and megavoltage therapeutic radiation
Lymphedema
Chemical exposure (2,3,7,8-etrachlorodibenzodioxin, Polyvinyl chloride
Parasitic infection (filariasis)
The majority of STS carry nonspecific genetic changes within a background of a complex karyotype. Approximately 15% to 20% of STS carry a specific translocation within a relatively simple karyotype which results in fusion proteins that can act as transcription factors, upregulating genes responsible for tumor growth, as in Ewing’s sarcoma, or translocate active promoters that regulate oncogenes which then lead to tumor formation (14). Other examples of sarcomas with specific genetic alterations with simple karyotypes include fusion genes due to reciprocal translocations and specific point mutations such as KIT mutations in GISTs and APC/β-catenin mutations in desmoid tumors. Examples of sarcomas with nonspecific genetic alterations and typically complex unbalanced karyotypes, representing numerous genetic losses and gains, include most types of adipocytic tumors that are characterized by specific chromosomal aberrations and reciprocal translocations, which can be diagnostically (15) and occasionally prognostically useful (16,17) (Table 52.2). The fusion gene translocations include 11 different gene fusions involving the EWS gene or EWS family members (TLS, TAF2N) found in five different sarcomas and ten other types of fusions in seven other sarcoma types (19). If conventional cytogenetics is not available, molecular genetic techniques (e.g., reverse transcription polymerase chain reaction and fluorescence in situ hybridization) are useful as diagnostic adjuncts. In addition, investigation of molecular changes of genes at the sites of chromosomal alterations has led to the identification of novel genes and the characterization of their mechanisms of deregulation. The tumor suppressor genes best studied in sarcoma are TP53 and RB1. Inactivation of both genes is involved in the tumorigenesis of several sarcomas. The relevance of the TP53 gene to sarcoma tumorigenesis is underscored by the frequent occurrence of STS in the Li-Fraumeni syndrome; all families studied have TP53 germline mutations. The major mechanisms of p53 pathway inactivation in sarcomas include p53 point mutations, homozygous deletion of CDKN2A, which encodes both p14ARF and p16, and HDM2 amplification. In sarcomas with specific reciprocal translocations, p53 pathway alteration is a rare event, but when present it is a strong prognostic factor, associated with significantly decreased survival in synovial sarcoma (20,21), myxoid liposarcoma (22), and Ewing’s sarcoma/peripheral neuroectodermal tumor (PNET) (18). Decreased survival in Ewing’s sarcoma/PNET was associated with deletion of CDKN2A, representing a type of p53 pathway alteration through loss of the CDKN2A alternative product p14ARF (23,24). In contrast, in sarcomas with nonspecific genetic alterations and complex karyotypes, p53 pathway alteration is more common and has weaker prognostic value, often requiring large numbers of patients to achieve statistical significance, as demonstrated in several studies of mixed adult STS. Its high prevalence in this class of sarcomas may account for its limited ability to define distinct clinical prognostic subsets in these tumors.
TABLE 52.2 CYTOGENETIC AND MOLECULAR ABNORMALITIES IN SARCOMAS
In addition to serving as very specific and powerful diagnostic markers, fusion genes resulting from translocations encode chimeric proteins that are important determinants of tumor biology, acting as abnormal transcription factors that alter the transcription of multiple downstream genes and pathways (25). The structure of these chimeric proteins play a prominent role in the pathogenesis of sarcoma, as evidenced by the impact of relatively minor cytogenetic variability, as a result of variant molecular breakpoints, on tumor phenotype and clinical behavior (17,26). A recent analysis of synovial sarcoma has clearly identified a characteristic SS18-SSX fusion gene resulting from the chromosomal translocation t(x; 18) (p11; q11) detectable in almost all synovial sarcomas. Translocation fuses the SYT gene from chromosome 18 to either of two highly homologous genes at Xp11, SSX1, or SSX2 and rarely SSX4. SS18-SSX1 and SS18-SSX2 fusion proteins are thought to function in aberrant transcriptional regulation. Recent analysis has suggested that these fusion products may influence outcome. It does appear that the majority of biphasic synovial sarcomas have an SS18-SSX1 fusion transcript, and most that were positive for SS18-SSX2 were monophasic. Conversely, monophasic sarcomas may have either transcript (16).
PATHOLOGIC EVALUATION
There are more than 50 histological subtypes of STS, many of which are associated with distinctive clinical, therapeutic, or prognostic features. Detailed descriptions of the histopathologic classification and guidelines for the histological reporting of soft tissue sarcoma have been published elsewhere (27). To summarize, the most commonly found are liposarcoma, MFH, and leiomyosarcoma. Histopathology is anatomic site dependent: the common subtypes in the extremity are liposarcoma or MFH; in the retroperitoneal/intraabdominal location, liposarcoma and leiomyosarcoma are the most common histiotypes, whereas in the visceral location, GIST and leiomyosarcoma are found almost exclusively. Liposarcoma is further classified into three major histological subtypes, based on strict morphologic features and cytogenetic aberrations: well-differentiated-dedifferentiated, myxoid-round cell, and pleomorphic (27). The well-differentiated-dedifferentiated subtypes account for 43% and 16% of liposarcoma, respectively, and are more commonly found in the retroperitoneal location. The myxoid/round cell and pleomorphic subtypes account for 29% and 12% of liposarcoma, respectively, and are usually located in the extremity. Multimodality treatment strategies aimed at the poorly differentiated malignant subtypes may include surgical resection, adjuvant radiation, and systemic chemotherapy, whereas surgical resection alone may be used in the welldifferentiated subtype, in particular in its lowest grade form, atypical lipoma (28). Age is also associated with specific histopathologies. In childhood, embryonal rhabdomyosarcoma is most common; synovial sarcoma is more likely to be seen in young adults (<35 years old); and there is an even distribution of liposarcoma and MFH as the predominant types in the older population (29). The designation MFH is currently being reevaluated with many of these tumors being reclassified as myofibrosarcoma, pleomorphic sarcoma, or dedifferentiated liposarcoma; a term increasingly used for these tumors is undifferentiated pleomorphic high-grade sarcoma (UPS). Sarcoma histiotype and liposarcoma subtype is generally an important determinant of prognosis and a predictor of distinctive patterns of behavior, because none of the existing grading systems is ideal and applicable to all tumor types. Biologic behavior is currently best predicted based on histological type, histological grade, tumor size, and depth. Although many published series have combined all the histological types of sarcoma, the significance of such subtyping is exemplified by liposarcoma in which the subsets (well differentiated, dedifferentiated, myxoid, round cell, and pleomorphic) have strikingly different biology and patterns of behavior (27,30,31,32). A further clear demonstration is the importance of myogenic differentiation in pleomorphic sarcomas, which is associated with a substantially increased risk of metastasis (33).
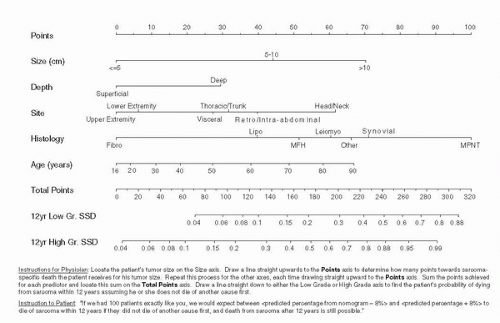
FIGURE 52.1. Kattan nomogram predicting the 12-year probability of sarcoma-specific death. (Adapted from Kattan MW, Leung DH, Brennan MF. Postoperative nomogram for 12-year sarcoma-specific death. J Clin Oncol 2002;20:791.)
In cases of poorly differentiated STS, immunohistochemical stains and molecular genetic techniques are often required to properly classify the tumor (34). For example, pleomorphic adult-type rhabdomyosarcoma will stain positive for desmin and myogenin (35). Even low-grade STS have a significant metastatic potential (9%) with death from distant disease associated with positive surgical margins, liposarcomatous histology, retroperitoneal tumor site, and less than a complete surgical resection (36). MSKCC investigators developed and internally validated a nomogram to predict the probability of a 12-year sarcomaspecific death using a database of 2136 patients. Nomogram variables included age, tumor size, histological grade, histological subtype, depth, and site (extremity, visceral, thoracic, trunk, retroperitoneal, head and neck). Sarcoma-specific death at 12 years was 36% and a boot strapped concordance index was 0.77 (Fig. 52.1). In this report, histological type was found to be one of the most important predictors of sarcoma-specific death, with malignant peripheral nerve sheath tumors having the highest risk of mortality (37).
This MSKCC nomogram was subsequently externally validated in collaboration with investigators from UCLA (1,167 STS patients) (38) and Milan, Italy (642 STS patients) in patients managed over a 20-year period (39). The MSKCC investigators also constructed a liposarcoma-specific nomogram that incorporated the five distinct histological types and increased the concordance index to 0.827 from 0.77, further highlighting the importance of histological subtype (40).
STS: CLINICAL PRESENTATION
Although STS can present in any part of the body, symptoms leading to a diagnosis vary. For example, STS arising in the extremities may present as a painless mass often considered by the patient to be pulled muscle or a posttraumatic bruise. STS arising in the retroperitoneum, pelvis, or viscera can cause nonspecific GI or GU symptoms and occasionally, due to massive size, a palpable abdominal mass. Retroperitoneal and visceral sarcomas account for about 34% of all sarcomas. The most common histopathological types in the retroperitoneum are liposarcoma (40%), leiomyosarcoma (25%), MPNST, and fibrosarcoma. In the visceral location GIST, leiomyosarcoma, and desmoids tumor are the most common histological types. About 55% of retroperitoneal liposarcomas will be well differentiated and low grade with roughly 40% of patients showing dedifferentiated, high-grade histology at primary presentation. Neurological symptoms related to retroperitoneal invasion or pressure on neurovascular structures can occur. Weight loss is uncommon and incidental diagnosis is often the norm. A delay in diagnosis is not uncommon for patients with STS. Unless subsequent imaging demonstrates unresectability, surgical exploration with complete resection is both therapeutic and diagnostic.
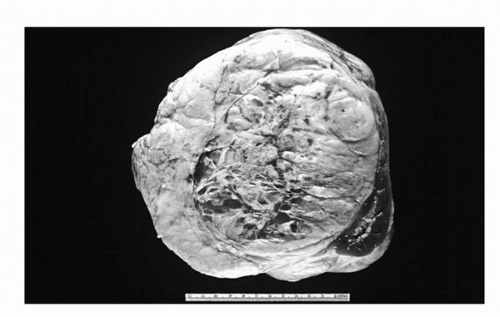
Occasionally massive benign tumors (i.e., angiomyolipomas) arising from the retroperitoneum or kidney can be clinically indistinguishable from STS and may require careful pathological, cytogenetic, and immunohistochemical analysis to define (Fig. 52.2). Important issues of differential diagnosis, particularly in the young, are the presence of a germ cell tumor, lymphoma, or a primary retroperitoneal tumor arising from the adrenal gland, which can often be distinguished by appropriate imaging (i.e., testicular ultrasound) and serological tumor markers (alpha feto protein, beta HCG). For patients with unresectable tumors or those for whom lymphoma is in the differential diagnosis, percutaneous CT-guided biopsy to obtain tissue is an appropriate first step prior to initiating systemic therapy (4). Depending upon the degree of suspicion, a diagnosis can be made by using a carefully placed needle biopsy or incisional biopsy after appropriate imaging. In >80% of patients undergoing a needle biopsy the tissue sample is adequate for diagnosis, and in >90% this biopsy will correlate with the final pathologic diagnosis obtained at the time of resection (4). In an analysis of 500 patients with retroperitoneal soft tissue sarcoma (41), median survival was 72 months for patients with primary presentation, 28 months for those with LR, and 10 months for those presenting with metastasis. For all patients with primary or locally recurrent STS, complete resection was the most favorable factor in outcome. Most patients present with an asymptomatic abdominal mass, although on occasion pain is present. Less common symptoms include gastrointestinal bleeding, incomplete bowel obstruction, and neurological symptoms related to retroperitoneal invasion or pressure on neurovascular structures.
FIGURE 52.2. Massive, benign angiomyolipoma with adjacent normal kidney resected from a 62-year-old female thought preoperatively to have a primary retroperitoneal sarcoma.
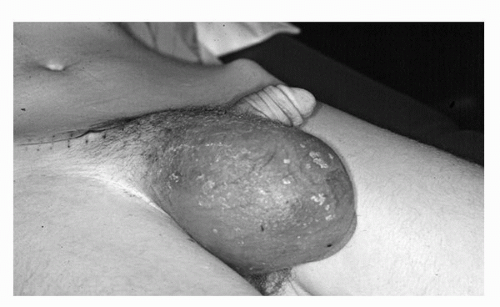
FIGURE 52.3. Massive paratesticular rhabdomyosarcoma in a 19-year-old.
GU STS PRESENTALTION
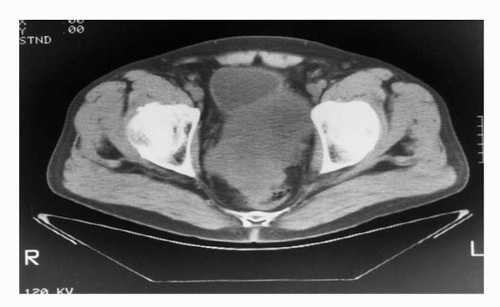
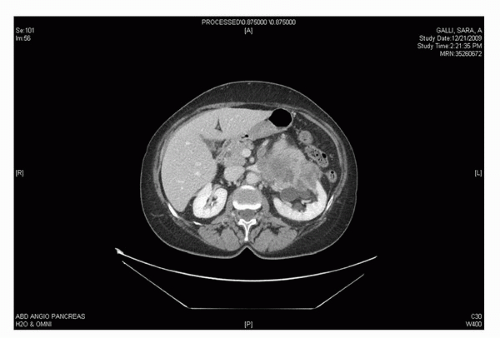
The presentation of GU STS ranges between those of the above-mentioned extremity STS and visceral retroperitoneal STS. For example, paratesticular STS can present as a painless groin mass and is often misinterpreted as a cord lipoma or inguinal hernia (6,42) (Fig. 52.3). Bladder STS, if involving the bladder lumen, can present with gross hematuria and irritative voiding symptoms and occasionally be diagnosed with a tumor small enough to be completely removed by transurethral resection (TUR). If the bladder STS growth is posterior into the pelvis rather than toward the bladder lumen, the lesion can reach massive proportions with direct extension into prostate, seminal vesicles, rectum, or the vagina and uterus in women. Such massive tumors can cause stranguria, abdominal cramping, obstipation, and bowel obstruction. STS from the prostate/seminal vesicle may also reach massive proportions with growth into pelvic and retroperitoneal spaces causing similar symptoms of stranguira, bladder outlet obstruction, urinary retention, rectal obstruction, or nonspecific abdominal discomfort. It is often difficult to determine whether the prostate, seminal vesicle, or bladder base is the origin of the tumor (Fig. 52.4). However, knowing the exact origin of the STS is not critical for formulation of the treatment plan. Kidney STS can present with pain, palpable mass, and gross hematuria as well as nonspecific GI complaints owing to massive size much in the same way retroperitoneal sarcomas present. Here too, it may be difficult to distinguish whether an STS tumor is arising from the kidney or the retroperitoneal soft tissues with secondary invasion of the kidney (43) (Fig. 52.5). Incidental detection of GU STS has also been reported during routine digital rectal exam associated with a yearly physical exam (seminal vesical sarcoma) (44) or during abdominal imaging for unrelated reasons (renal angiosarcoma) (45) (Fig. 52.6).
FIGURE 52.4. CT scan demonstrates pelvic high-grade leiomyosarcoma with difficulty distinguishing a primary arising from the prostate or seminal vesical in a 62-year-old male.
FIGURE 52.5. Perirenal high-grade leiomyosarcoma invading kidney in a 62-year-old female.
EXTENT OF DISEASE EVALUATION
All patients require a thorough history and physical examination. Anatomic imaging with plane radiographs, computed tomography (CT), and magnetic resonance imaging (MRI) can all play a role in the initial evaluation of STS. FDG positron emission tomography (PET) and newer radiopharmaceuticals, such as 18F-fluorodeoxythymidine, can be used to evaluate distant metastases and assess whether a lesion is a benign or low-grade STS (46). MRI examination is usually the preferred imaging study for STS since it enhances the contrast between tumor and adjacent vascular, neural, or visceral structures and provides excellent definition of fascial and neurovascular planes. Once the diagnosis and grade of the STS are known, evaluation for sites of potential metastasis can be performed. Lymph node metastases occur in <3% of adult soft tissue sarcoma (47). For extremity lesions, the lung is the principal site for metastasis of high-grade lesions whereas for visceral lesions, the liver is the principal site of metastasis (48). Thus, patients with low-grade extremity lesions require a chest radiograph, and the majority of those with high-grade lesions require a chest CT. Patients with visceral lesions should have their liver imaged as part of the initial abdominal CT or MRI. Routine bone scans and head CT or MRI are not ordered in the absence of localizing symptoms. For patients with bladder and prostate/seminal vesical STS, examination under anesthesia (EUA) is a critical maneuver to determine whether a mass is fixed to the pelvic side walls or rectum. Cystoendoscopy with or without proctosigmoidoscopy may be performed to determine direct extension in these organs. Consulting colorectal or general surgeons should be present for the EUA in order to optimize surgical planning. For patients with small bladder STS presenting with gross hematuria, aggressive TUR may be both diagnostic and therapeutic (6). For massive tumors that are clinically unresectable, percutaneous needle biopsy is recommended and effectively provides sufficient tissue for an accurate diagnosis in the vast majority of cases.
Only gold members can continue reading. Log In or Register to continue
 Androgen Receptor Signaling in Castration-Resistant Prostate Cancer
Androgen Receptor Signaling in Castration-Resistant Prostate Cancer



