ADRENOCORTICAL INSUFFICIENCY
PRIMARY FORM
ETIOLOGY
Congenital Adrenal Hyperplasia and Hypoplasia.
The most prevalent cause of adrenocortical insufficiency in infancy is congenital adrenal hyperplasia (Fig. 83-1). This condition is discussed extensively in Chap. 77. Further details are emphasized in the sections Treatment and Precocious Pseudopuberty.
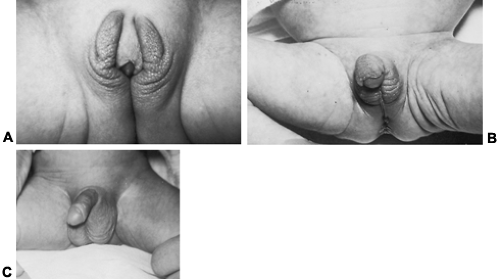 FIGURE 83-1. Congenital adrenal hyperplasia in three newborn infants, all with 21-hydroxylase deficiency. The female infants depicted in A and B have different degrees of ambiguity of the external genitalia. A, Clitoromegaly with labial enlargement. B, Severe clitoral hypertrophy and nearly complete labial fusion. Note the scrotal appearance of the labia. C, Male infant with precocious development of the external genitalia and rapid somatic growth (infant Hercules). (Courtesy of Dr. Wellington Hung.) |
By contrast, congenital adrenal hypoplasia is a rare cause of adrenal insufficiency in infancy; it has been reported to occur as an apparently isolated defect, with or without a familial tendency.9 The autosomal recessive form is associated with miniature, adult-type, adrenal architecture and may be secondary to isolated adrenocorticotropic hormone (ACTH) deficiency. The X-linked form is due to DAX-1 gene mutations and is associated with cytomegaly of fetal-like adrenocortical cells and congenital gonadotropin deficiency.10 Mental retardation and other features may occur, depending on the extent of contiguous chromosomal deletion.
Neonatal Adrenal Hemorrhage.
Neonatal adrenal hemorrhage usually presents with the symptoms of shock, severe jaundice, and an abdominal mass during the first week of life.11 It is associated with birth trauma and is thought to result from severe hypoxemia. Ultrasonographic examination, repeated at 3- to 5-day intervals, may reveal a lesion that changes from solid to cystic as the hemorrhagic mass liquefies, and adrenal calcifications may be noted after 4 weeks of age. Children who survive should be tested for chronic adrenal insufficiency.
Destruction of the Adrenal Cortex.
Beyond the newborn period, adrenal insufficiency usually is attributable to destruction of the adrenal cortex. Addison disease usually is caused by autoimmune endocrinopathy that may be associated with diseases affecting other glands (see Chap. 76 and Chap. 197). The major autoantigens are the steroidogenic enzymes, particularly 21-hydroxylase (cytochrome P450c21),12 but their presence does not necessarily predict adrenal failure.13 There are two varieties of autoimmune polyglandular deficiency syndrome. Type I usually is seen in early life, with a mean age at onset of 12 years. Although Addison disease in this syndrome is commonly associated with hypoparathyroidism and mucocutaneous candidiasis, the incidence of associated type 1 diabetes mellitus and thyroid disease is low.14 Pernicious anemia and other autoimmune disorders are seen occasionally. The disorder is an autosomal recessive disease due to mutation of an autoimmune regulator gene on chromosome 21q22.3.15 Type II autoimmune polyglandular deficiency has a mean age at onset of 30 years. This form of the disease is associated with insulin-dependent diabetes mellitus, thyroid deficiency, or both, and has an autosomal dominant transmission pattern linked with the human leukocyte antigen (HLA)-B8 allele. Both type I and type II can be associated with vitiligo and primary gonadal failure.
Acute adrenal insufficiency can complicate septic shock. The classic association of acute adrenal hemorrhage and purpura with fulminating meningococcemia (Waterhouse-Friderichsen
syndrome)16 (Fig. 83-2; see Chap. 213). It resembles the generalized Shwartzman phenomenon.
syndrome)16 (Fig. 83-2; see Chap. 213). It resembles the generalized Shwartzman phenomenon.
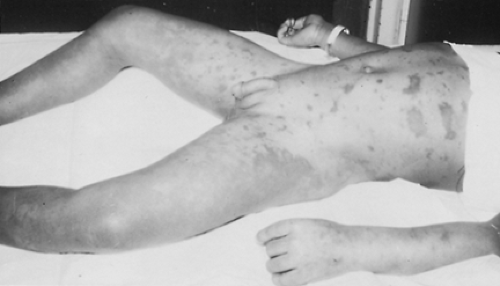 FIGURE 83-2. Seven-year-old boy with Waterhouse-Friderichsen syndrome secondary to severe menin-gococcemia. The child subsequently died. Note the massive hemorrhaging into the skin. (Courtesy of Dr. Wellington Hung.) |
Progressive degenerative or granulomatous diseases such as tuberculosis or histoplasmosis may involve the adrenal cortex and cause adrenal failure. Lysosomal acid lipase deficiency, or Wolman disease, is a disorder that occurs in infancy. It is caused by abnormal storage of triglycerides and cholesterol esters that cannot be degraded by the adrenals, spleen, liver, bone marrow, brain, or lymph nodes. The form of transmission is autosomal recessive.17 Patients present in the early weeks of life with vomiting, diarrhea, failure to thrive, hepatosplenomegaly, and adrenal calcification. Invariably, the patient progresses to death by a few months of age despite corticosteroid treatment.
Adrenoleukodystrophy is a rare, fatal, or autosomal recessive demyelinating disorder of the brain that presents in childhood with blindness, quadriparesis, or dementia, typically before adrenal failure.18,19 Adrenomyeloneuropathy, a more slowly progressive form, may present in boys as adrenal failure. These disorders result from accumulation of very long-chain fatty acids (C-24 and longer). Most forms are X-chromosome–linked, due to mutations of a gene encoding a peroxisomal integral membrane protein that is located on Xq28. Neonatal adrenoleukodystrophy is an autosomal recessive disease caused by defects in peroxisome assembly; it results from mutations of PEX genes.19
Isolated Glucocorticoid Insufficiency.
Glucocorticoid deficiency in the presence of normal mineralocorticoid secretion can result from primary adrenal disease or be secondary to defects in the secretion or mode of action of ACTH. In the early stages of autoimmune Addison disease, aldosterone secretion is sometimes
intact. This must be distinguished from ACTH deficiency, which has been reported in the polyglandular autoimmune syndrome.20
intact. This must be distinguished from ACTH deficiency, which has been reported in the polyglandular autoimmune syndrome.20
Familial isolated glucocorticoid deficiency appears to be caused by a heterogeneous group of autosomal recessive disorders. These include degenerative disorders of the zona fasciculata and defects in the ACTH receptor, or ACTH action.21 ACTH resistance may result from degeneration of the zona fasciculata-reticularis in association with achalasia of the esophagus and alacrima (triple-A syndrome).21 Approximately 15% of patients later develop mineralocorticoid deficiency. This disorder maps to a critical region on the chromosome 12q13 locus. Patients with isolated ACTH receptor defect average 2 standard deviations (SD) taller than average.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


