FIGURE 104-1. Combined fetal adrenal weights according to gestational age.
(Data from Young MC, Laurence KM, Hughes IA: Relationship between fetal adrenal morphology and anterior pituitary function, Horm Res 32:130–135, 1989.)
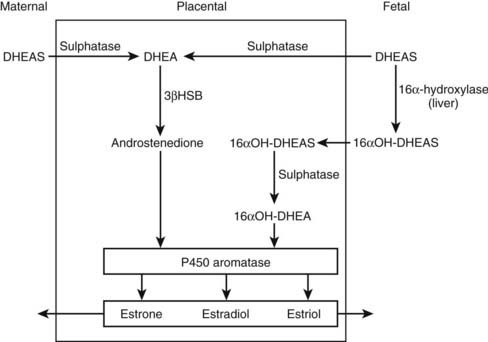
In concert with the morphologic changes in the fetal adrenal, there is a vast output of steroids in the form of DHEA, pregnenolone, 170H-pregnenolone, and their respective sulfated conjugates. The steroid output amounts to about 200 mg per day, of which 60% is DHEAS.4 This C19 fetal steroid is a major substrate for estrogen biosynthesis via the fetoplacental unit (Fig. 104-2). Key enzymes are 16α-hydroxylase to form 160H-DHEAS in the liver, placental sulfatase to produce free DHEA, and the action of placental P450 aromatase to synthesize the estrogens, estrone, estradiol, and estriol, from their respective C19 androgen substrates. The abundance of C19 steroids, which are normally aromatized by the placenta, is illustrated by the profound virilizing effects on the mother and her female fetus in the presence of an inactivating mutation of the CYP19 gene.5
POSTNATAL ADRENAL MORPHOLOGY AND FUNCTION
A precipitous fall in adrenal weight by about 50% occurs after birth as a result of involution of the fetal zone. The definitive zone remains and is the template for development into the characteristic zones of the adult adrenal cortex. It is proposed that a combination of proliferation and migration of progenitor cells underlies this characteristic tissue zonation.6 The rapid reduction in adrenal gland size within weeks of birth is an apoptotic process rather than due to hemorrhage or necrosis.7 It appears to be independent of ACTH and is induced by the action of activin and transforming growth factor alpha (TGF-α). Involution of the fetal zone is reflected biochemically in a concomitant decrease in DHEAS levels, which remain low until the increase characteristic of adrenarche from ages 6 to 8. There is thickening of the zona reticularis evident by age 3.8 Longitudinal studies of 24-hour urinary androgen excretion and serum DHEAS concentrations suggest there is already a gradual increase in C19 steroid output from the postnatal adrenal gland from early childhood.9,10 This recrudescence of the pattern of fetal adrenal steroidogenesis indicates a common mechanism involving modulating factors operating within the adrenals to increase C19 steroid production independent of ACTH.
MECHANISMS OF ANDROGEN SECRETION AT ADRENARCHE
DHEAS is the most abundant endogenous steroid produced within the endocrine system and circulates in micromolar concentrations. The pattern of secretion throughout prenatal and postnatal life is depicted in Fig. 104-3, and the prerequisites for DHEA and DHEAS synthesis are shown in Fig. 104-4.
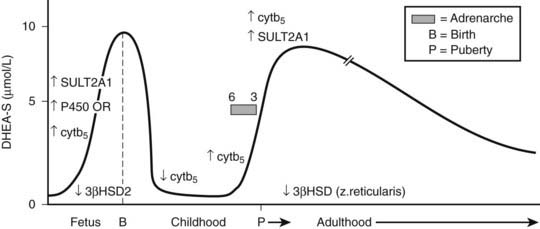
FIGURE 104-3. Schematic of dehydroepiandrosterone sulfate (DHEAS) production from fetal to adult life. The arrows indicate temporal changes in factors controlling DHEAS (see text).
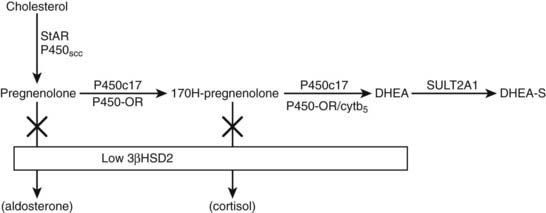
FIGURE 104-4. Pathway of dehydroepiandrosterone sulfate (DHEAS) synthesis in fetal adrenal and adult zona reticularis. Also depicted is the effect of low expression of 3β-hydroxysteroid dehydrogenase 2 (3β-HSD2) in these tissues.
The initial step in converting cholesterol to pregnenolone is common to all steroid-producing glands and is rate limiting.11 The key steroidogenic junction is at the step mediated by the P450c17 enzyme and defined as the qualitative regulator of steroidogenesis.12 This enzyme uniquely performs two catalytic activities called 17α-hydroxylase and 17,20-lyase. It is the latter enzyme activity that preferentially ensures C19 steroid production and, in particular, DHEA synthesis and adrenarche. A number of factors enhance the differential activities of P450c17. These include posttranslational regulation of P450c17 by phosphorylation on serine/threonine residues and the role of electron transfer proteins.13–15 The principal electron donor to microsomal P450 enzymes, including P450c17, is nicotinamide adenine dinucleotide phosphate (NADPH) cytochrome P450 oxidoreductase. This enzyme cofactor augments both 17α-hydroxylase and 17,20-lyase activities but is not the principal determinant of increased DHEA synthesis. However, mutations in the P450 oxidoreductase gene lead to a biochemical profile reminiscent of combined partial 17α-hydroxylase and 21-hydroxylase deficiencies with virilization in affected girls and undermasculinization in affected boys.16,17 The Antley-Bixler syndrome, a condition characterized by severe skeletal abnormalities, genital anomalies, and adrenal dysfunction, is also a manifestation of this enzyme deficiency.18 Enhancement of 17,20-lyase activity is linked specifically to the action of cytochrome b5 in the presence of adequate P450 oxidoreductase.12 This effect is far more substantial with Δ5 substrates such as 170H-pregnenolone as compared with a Δ4 substrate such as 170H-progesterone. Hence, the predominance of DHEA synthesis. Coupled to the promotion of DHEA synthesis is a relative deficiency of 3β-hydroxysteroid dehydrogenase (HSDB2) in the fetal adrenal and postnatal zona reticularis. This enzyme would normally compete with P450c17 to convert Δ5 to Δ4 steroids.19
More than 99% of DHEA is sulfated to DHEAS through the action of dehydroepiandrosterone sulfatase (SULT2A1).20 Sulfated steroids are also unavailable as substrates for HSDB2 activity, thus maintaining the high levels of DHEAS. Immunohistochemical studies in the fetal adrenal and in age-related postnatal adrenal glands have demonstrated a developmental pattern of enzyme and cofactor expression that may explain the intraadrenal control of adrenarche.21–23 In summary, the pattern of increased DHEAS secretion is associated with increased expression of P450c17, P450 oxidoreductase, cytochrome b5, SULT2A1, and decreased expression of HSDB2 in the fetal adrenal and again in the postnatal zona reticularis after age 5. In contrast, a reversal of this pattern occurs in the postnatal adrenal gland between infancy and age 5. Expression of cytochrome b5 and SULT2A1 is maintained beyond the adult age when DHEAS levels start to decline. The zona reticularis becomes smaller with increasing age, suggesting that the decline in DHEAS levels is associated with a decrease in cell number rather than a change in enzyme content. What factors stimulate the changes in expression of key proteins controlling DHEAS synthesis remain to be determined. HSDB2 has the important function of determining the ratio of Δ5 and Δ4 steroid production. A family of orphan nuclear receptors (nerve growth factor–induced clone B [NGF1B]) is expressed in parallel with HSDB2 and directly act on the promoter to up-regulate the HSD3B2 gene.24 It is possible that decreased expression of this and other transcriptional regulators of HSD3B2 at key developmental stages allows the increased production of DHEAS during fetal life and at adrenarche.
EXTRAADRENAL CONTROL OF ADRENARCHE
There is sufficient dissociation between changes in cortisol and DHEAS secretion to indicate that ACTH is not the primary trophic factor responsible for the rise in DHEAS levels at adrenarche. Nevertheless, ACTH may play a permissive role because DHEAS levels are either undetectable or significantly decreased in familial glucocorticoid deficiency associated with ACTH resistance.25 Corticotropin-releasing factor (CRH) has been suggested as a specific adrenal androgen secretagogue in view of the direct effect of CRH on human fetal adrenal cells in stimulating DHEAS secretion and following CRH infusion in normal men.26,27 However, the interpretation of such in vivo data is compounded by the concomitant effect of increased ACTH secretion.
Numerous other factors have been postulated to have effects on adrenal androgen secretion, mainly based on in vitro studies using isolated human fetal adrenal cells. These include prolactin, estrogens, TGFs, cytokines, insulin-like growth factors (IGFs), and a fragment of pro-opiomelanocortin (POMC). None has proven to be a specific adrenal androgen secretagogue. Leptin differentially regulates 17,20-lyase activity in an adrenocortical carcinoma cell line that expresses the leptin receptor.28 There is also an association between DHEAS and leptin levels at the time of adrenarche in obese children.29 Overall, the adrenarche results from an alteration of expression of CYP17, HSD3B2, CYB5 (cytochrome b5), and SULT2A1, intertwined with changes in protein kinase A and insulin signaling pathways.
CLINICAL FACETS OF ADRENARCHE
The term adrenarche defines the rise in adrenal androgens (DHEA, DHEAS, and androstenedione) that occurs between 6 and 8 years of age as a result of the modulation of adrenal steroidogenesis previously described; it is thus a biochemical definition for an event that is not usually translated into clinical signs. Adrenarche is often used interchangeably with pubarche, which describes the growth of sexual hair in the suprapubic area and axillae. Adrenal androgens are the stimuli for public and axillary hair growth in females, and hair growth typically starts on the labia. In boys, the distinction is less clear because the growth of pubic and axillary hair is primarily stimulated by the rise in testicular testosterone production. Adrenarche (or pubarche) is considered premature when there is onset of pubic hair growth before age 8 in girls and before age 9 in boys. Because all children have an increase in adrenal androgen production between 6 and 8 years of age, it remains perplexing why hair growth is expressed only in a small minority. Premature adrenarche is more common in girls than boys, and there is an association with low birth weight.30 However, this sex dimorphism is not apparent in normal children in whom a relationship between lower birth weight and higher adrenal androgen levels is present in both sexes.31 Children with premature adrenarche are typically taller than average, may have increased body odor and acne, and exhibit a bone age advanced by 1 to 2 years. In a study of girls with premature adrenarche, increased linear growth was already evident in the first 2 years of life and associated with higher IGF-1 levels.32 The taller stature later is probably the result of increased androgen levels, yet the mini–growth spurt in normal children that also occurs around ages 6 to 8 is probably not a function of adrenarche.33 Obesity is generally associated with taller stature in childhood, and the body mass index (BMI) tends to be higher in premature adrenarche, but the BMI does not correlate with androgen levels.29
Table 104-1 lists the conditions that should be excluded when assessing a child with early pubic hair development and the appropriate investigations to perform in such cases. Imaging using ultrasound or computed tomography (CT) will readily exclude an adrenal tumor. Furthermore, if the cause is an adrenal tumor, signs of virilization are more profound. These signs include clitoromegaly, hirsutism, voice deepening, and in boys, penile growth.34 The main condition to exclude is late-onset congenital adrenal hyperplasia (CAH) which is found in about 5% of children with early onset of pubic hair. Baseline measurements of 170H-progesterone, androstenedione, testosterone, and DHEAS are usually sufficiently sensitive and specific to distinguish the two conditions.35 Further confirmation rests with mutational analysis of the CYP21 gene. Premature adrenarche has generally been regarded as a benign variant with a normal age of menarche in girls and no negative effect on adult height.36 However, more detailed longitudinal studies of premature adrenarche suggest an increased risk of later ovarian hyperandrogenism in adolescent girls and hyperinsulinism.37 It is possible that premature adrenarche is a risk factor for later development of the metabolic syndrome in adult life. Daughters of mothers with polycystic ovary syndrome are at higher risk of developing this syndrome, and a significant number exhibit exaggerated adrenarche.38 The role of sulfotransferases in providing adrenal substrates for androgen production is vividly illustrated by a child with premature adrenarche who subsequently progressed to features of the polycystic ovary syndrome.39 Investigations showed compound heterozygous mutations in the gene encoding for a sulfate donor required for sulfation of DHEA to DHEAS by DHEA sulfotransferase. The net result is an increase in unconjugated DHEA, which provides the substrate for increased adrenal androgen production. Currently, there is no indication that the use of insulin sensitizers should be considered in a child presenting with typical clinical and biochemical features of premature adrenarche, but it is apposite to counsel parents that affected girls may later experience menstrual disturbances in adolescence. Early intervention after menarche with insulin sensitizers may prevent progression to polycystic ovarian syndrome and reduce the risk of long-term cardiovascular problems.40
Table 104-1. Causes of Early Pubic Hair and Relevant Investigations
ROLE OF ADRENAL ANDROGENS IN ADULTS
The physiologic role of adrenal androgens, including DHEAS, is not well understood. The association of adrenarche with pubic and axillary hair development suggests a role for DHEAS as a substrate for androgen synthesis. There is evidence that DHEAS is converted to sex steroids (testosterone, estradiol) in peripheral tissues via several enzymatic steps. Circulating, adrenally derived DHEAS is first converted to DHEA by sulfatase, which is further converted to androstenedione by 3β-hydroxysteroid dehydrogenase (3β-HSD); in turn, androstenedione is modified to testosterone or estradiol by isozymes of 17β-HSD or P450 aromatase, respectively (Fig. 104-5). It has been suggested that such conversion, occurring intracellularly via these enzymes that are widely expressed in many peripheral tissues, constitutes an intracrine mechanism for local generation of sex steroids from DHEA. This mechanism might also account for the observation that DHEA administration can result in androgenic/estrogenic effects without significant changes in circulating levels of these steroids.41 There is also evidence to suggest that DHEA can act as a neurosteroid to exert effects in the central nervous system. Steroidogenic enzymes mediating DHEA synthesis are expressed in the brain, and it has effects on neuronal growth and differentiation.42 DHEA has also been shown to modulate neurotransmitter signaling, acting as an allosteric antagonist at the γ-aminobutyric acid (GABA) receptor but as an agonist at the N-methyl-d-aspartate (NMDA) receptor.43 DHEA may have a further potential role as an antiglucocorticoid. It has been shown to antagonize glucocorticoid-induced thymic involution or corticosterone-mediated neurotoxicity to hippocampal cells.44,45
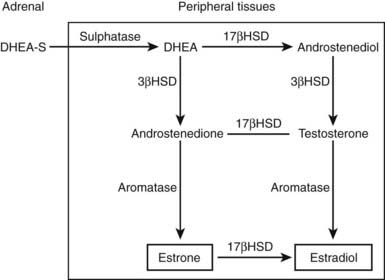
FIGURE 104-5. Pathways by which dehydroepiandrosterone sulfate (DHEAS) is converted intracellularly to sex steroids (testosterone, estradiol) in peripheral target tissues. Elevation in levels of androgen metabolites (e.g., androstanediol glucuronide) following DHEA treatment provides evidence for such peripheral sex steroid synthesis.
To date, a specific receptor that mediates the action of DHEA has not been identified, and whether it acts at the cell surface or intracellularly remains to be elucidated. DHEA has been shown to stimulate nitric oxide synthesis in endothelial cells via a specific G protein–coupled membrane receptor46 and to activate the mitogen-activated protein kinase (MAPK) pathway in vascular smooth muscle cells independently of androgen and estrogen receptors.47 Microarray studies of human peripheral blood mononuclear cells indicate that DHEA induces a gene expression profile that is distinct from glucocorticoid and testosterone, supporting the notion that it may act via an independent pathway.48
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


