AML with recurrent genetic abnormalities
AML with t(8;21)(q22;q22.1); RUNX1-RUNX1T1
AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22);CBFB-MYH11
APL with PML-RARA
AML with t(9;11)(p21.3;q23.3); MLLT3-KMT2A
AML with t(6;9)(p23;q34.1); DEK-NUP214
AML with inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2); GATA2, MECOM
AML (megakaryoblastic) with t(1;22)(p13.3;q13.3); RBM15-MKL1
AML with mutated NPM1
AML with biallelic mutations of CEBPA
Provisional entity: AML with BCR-ABL1, AML with mutated RUNX1
AML with myelodysplasia-related changes
Therapy-related myeloid neoplasms
AML, not otherwise categorized (NOS)
AML with minimal differentiation
AML without maturation
AML with maturation
Acute myelomonocytic leukemia
Acute monoblastic/monocytic leukemia
Pure erythroid leukemia
Acute megakaryoblastic leukemia
Acute basophilic leukemia
Acute panmyelosis with myelofibrosis
Myeloid sarcoma
Myeloid proliferations related to Down syndrome
Transient abnormal myelopoiesis (TAM)
Myeloid leukemia associated with Down syndrome
3.3 Treatment of De Novo AML
The principle of AML treatment is based on “total cell kill” theory, to eradicate all the leukemic blasts that have expanded inside the patient’s body. This is attempted by multi-agent chemotherapy with optional use of hematopoietic stem cell transplantation (SCT) for high risk of relapse. In addition, risk stratification, means of optimizing therapy by evaluating relapse risk with known prognostic factors, and supportive care have contributed to improve survival rate in children with AML.
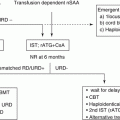
Treatment algorithm of pediatric AML proposed in the guideline of the Japanese Society of Pediatric Hematology and Oncology is shown in Fig. 3.1. ML-DS and APL are treated separately with each disease-specific protocol. The rest of the children with AML (de novo AML) are treated with appropriate risk-stratified therapies.

Fig. 3.1
Algorithm of treatment for children with acute myeloid leukemia. AML, acute myeloid leukemia; APL, acute promyelocytic leukemia; ATRA, all-transretinoic acid; CR, complete remission; DS, Down syndrome; HSCT, hematopoietic stem cell transplantation. [Adopted from Clinical Guideline for Pediatric Leukemia and Lymphoma (ver.3) Japanese Society of Pediatric Hematology and Oncology.]
3.3.1 Prognostic Factors
3.3.1.1 Biologic and Genetic Features
It is considered that AML occurs as a consequence of malignant transformation of an abnormal single myeloid progenitor cell via multi-step alterations. One model classified the different types of these mutations in two types: the Type I mutations including mutations that function as proliferation and survival signals (e.g., FLT3, KIT, RAS) and the Type II mutations including mutations that lead to differentiation arrest or enhanced self-renewal (e.g., RUNX1-RUNX1T1, CBFB-MYH11) [12]. The leukemia stem-cell model proposes that the clonal origin of AML cells reside at the top of hierarchical organization, possessing unlimited self-renewal capacity and capability of propagating leukemia cells [13]. Recent genome-wide analyses are challenging to uncover full landscape of AML leukemogenesis. These novel technologies have found that AML contains fewer genetic alterations than do other malignancies. In particular, pediatric AML contains only 2–4 somatic copy-number alterations per single leukemia cell and no copy-number alterations in approximately one-third of cases, indicating that the development of AML may require fewer genetic alterations compared to other malignancies [14]. Although full picture of AML leukemogenesis is still unclear, disease-specific gene alterations regulate biological character of AML cells, thus have prognostic value, and would be good candidates for therapeutic target.
- (a)
Core-Binding Factor AML
AML with t(8;21) or either inv(16) or t(16;16) are called “core-binding factor (CBF)” AML and are associated with favorable prognosis. t(8;21) creates RUNX1-RUNX1T1 fusion gene. The abnormal fusion protein binds DNA and interacts with CBFβ, but dominantly represses transcriptional activation through interactions with the nuclear corepressor complex. t(8;21)-AML comprises 12% of pediatric AML in the USA and Europe and higher prevalence in east Asia (nearly 30% of pediatric AML in Japan). It is clinically associated with FAB M2 morphology and extramedullary disease. Large international retrospective analysis of 838 patients with t(8;21)-AML by the International BFM Study Group (I-BFM-SG) showed additional del(9q) and +4 were associated with treatment failure [15]. Additionally, patients with t(8;21)-AML were likely to benefit from protocols that included high doses of anthracyclines, etoposide, and cytarabine.
inv(16) or t(16;16) creates CBFB-MYH11 fusion gene. The rearrangement causes formation of abnormal CBFβ with impaired function, thus leading to improper differentiation of myeloid cells. AML with inv(16) accounts for 10% or less of pediatric AML. It is associated with myelomonocytic morphology with abnormal eosinophilia (FAB M4Eo).
The KIT gene, encoding a type III receptor tyrosine kinase, is mutated in nearly 20% of CBF-AML. Prognostic impact of mutated KIT in CBF-AML is controversial [16–18]; however, recent analysis among the Japanese AML-05 cohort showed significant poor prognosis of KIT exon 17 mutations in t(8;21)-AML; especially exon 17 D816V mutation was associated with higher relapse rate [19].
- (b)
KMT2A Gene Rearrangements
KMT2A (MLL) gene, located at chromosome band 11q23, encodes a histone methyltransferase that is involved in epigenetic regulation of blood cell development [20]. Rearrangements of MLL occur as a result of balanced chromosomal translocations that fuse the MLL gene to one of more than 70 known partner genes [21]. MLL rearrangement accounts for 15–20% of pediatric AML, and the most common is MLL-AF9 derived from t(9;11)(p22;q23) which accounts for ~50% of the cases. International retrospective analysis of 756 children with MLL-rearranged AML by the I-BFM-SG identified large differences in outcome according to MLL subtypes, showing significant poor survival rate of t(1;11)(q21;q23), t(6;11)(q27;q23), t(10;11)(p12;q23), and t(10;11)(p11.2;q23) cases [22]. Recent analysis of the Japanese AML-05 cohort showed clear difference in the outcome of MLL-AF9 AML according to the expression of the ecotropic viral integration site-1 (EVI1) gene, overexpression being a poor prognostic factor [23].
- (c)
Genetic Alterations in Non-Down Syndrome Acute Megakaryoblastic Leukemia
AMKL (FAB M7) accounts for 10% of AML in non-Down syndrome (non-DS) children. Unlike DS AMKL with mutated GATA1, prognosis of non-DS AMKL is reported to be poor [24], but the outcome is moderately improving with modern intensive chemotherapy [25]. Heterogeneity of non-DS AMKL has been described in several studies. I-BFM-SG reported that non-DS AMKL could be classified in three groups: good risk group with 7p abnormalities; poor risk group with normal karyotypes, −7, 9p abnormalities including t(9;11)/MLL-AF9, −13/13q-, and −15; and intermediate risk group with others including t(1;22)/RBM15-MKL1, found in infants, and 11q23/MLL rearrangements other than MLL-AF9 [26]. Recent genomic studies identified two novel recurrent genetic abnormalities in non-DS AMKL: CBFA2T3-GLIS2 created by cryptic inv(16)(p13.3q24.3) and NUP98-KDM5A (formerly known as NUP98-JARID1A) by cryptic t(11;15)(15;q35) [27, 28]. CBFA2T3-GLIS2 and NUP98-KDM5A comprise ~15 and ~10% of pediatric non-DS AMKL cases, respectively, and both confer poor prognosis [29].
- (d)
FLT3-ITD
FLT3-ITD accounts for more than 20% of adult AML and 5–10% of pediatric AML and is associated with poor prognosis [30–32]. FLT3-ITD is most commonly observed in cytogenetically normal (CN)-AML, but with other translocations as well. It is considered that the presence of FLT3-ITD has no prognostic impact when present with favorable risk cytogenetics such as CBF-AML or t(15;17)/PML-RARA APL. Some study groups such as Children’s Oncology Group (COG) include only FLT3-ITD with high allelic ratio in high-risk criteria.
- (e)
NUP98-NSD1
Nucleoporin 98kD (NUP98) gene located on chromosome 11p15 fuses to more than 20 different partner genes. Among them, NUP98-NSD1 created by cryptic t(5;11)(q35;p15.5) was recently identified by the Dutch group using array-based genomic tests [33]. NUP98-NSD1 comprised 4.2% of pediatric AML and 16.1% of CN-AML. Clinically, this abnormality is associated with FAB M4 and M5 and the presence of FLT3-ITD and WT1 mutations and confers poor prognosis.
- (f)
Others
Monosomy 7 and monosomy 5 or 5q deletions account for ~5% of pediatric AML and are traditionally used as poor-risk factors in many studies [34, 35]. Monosomy 5 or 5q deletions are associated with complex karyotype (>3 chromosomal abnormalities, excluding recurrent changes). Other poor prognostic cytogenetics include t(16;21)(p11;q22)/FUS-ERG, t(9;22)/BCR-ABL1, t(6;9)/DEK-NUP214, and inv(3) or t(3;3)/RPN1-MECOM, but are very rare in children.
With conventional cytogenetic analysis, approximately 20% of the cases fall into a CN-AML category with intermediate prognosis. However, development of genetic studies has revealed that CN-AML is a heterogeneous disease with various prognoses. Among them, FLT3-ITD and NUP98-NSD1 are associated with unfavorable outcome as previously described, while biallelic mutations of CEBPA and NPM1 are associated with favorable outcome [36–40].
3.3.1.2 Early Treatment Response
Risk stratification evaluating morphological treatment response in the bone marrow after the first course of chemotherapy is currently utilized in many pediatric AML study groups, which is predictive of final outcome.
Recently, measurement of minimal residual disease (MRD) targeting leukemic clone-specific features has been increasingly used for better risk stratification. Methods that are applicable to AML are flow cytometry detecting aberrant combinations of leukemic cell-surface antigen, polymerase chain reaction (PCR) amplification of leukemia-specific mutated gene transcripts, quantitative measurement of WT1 expression, etc. Detection of fusion transcripts by reverse transcript (RT)-PCR method has sensitivity of 10−4–10−5; however, it is applicable to only 50% of the cases. Moreover, it is well known that fusions such as RUNX1-RUNX1T1 and CBFB-MYH11 could persist in patients with long-term remission. Hence, flow-based MRD is more widely used because of its applicability (more than 90% of the cases) and its specificity, although the sensitivity is one log lower than the PCR-based method. In the AML02 study of St. Jude Children’s Research Hospital, MRD of 1% or higher after initial induction course clearly segregated patients with high and low risk of relapse, and the relapse rate was only 17% for the patients whose MRD level was below 0.1% after the second induction [41]. However, both disease-free survival (DFS) and OS rates were low for poor prognostic patients [e.g., t(6;11), t(10;11), AMKL without t(1;22)] even with negative MRD (<0.1%) [42], suggesting the stronger impact of biological factors on outcome in AML.
3.3.2 AML Therapy
Therapy for AML is consisted of remission-induction and post-remission phases. In both phases, chemotherapy including two major key drugs, cytarabine and anthracyclines, is the mainstay of the treatment. Choice of post-remission therapy, especially indication of allogeneic SCT, is determined by risk of relapse evaluated by known prognostic factors.
3.3.2.1 Remission-Induction Phase
Remission-induction therapy aims to achieve morphological complete remission (CR), defined as less than 5% blasts in bone marrow with regeneration of normal hematopoiesis and no evidence of residual extramedullary disease. Time of evaluating remission differs by protocol, but most typical is after completing two courses, and 80–90% of the children achieve remission in contemporary therapy.
The first successful induction regimen was “3 + 7” established in 1980s by the Cancer and Leukemia Group B (CALGB) in the USA for adult AML, 7 days of cytarabine (100 mg/m2) by continuous infusion plus 3 days of daunorubicin (45 mg/m2), which resulted in 60–70% CR rate [43]. Until now, intensification of “3 + 7” has been attempted to improve the outcome of AML.
- (a)
Cytarabine Intensification
CALGB 8321 study compared cytarabine dose of 200 mg/m2/day with 100 mg/m2/day in induction, but could not find clear advantage of higher cytarabine dose [44]. Still, several studies use cytarabine dose of 200 mg/m2: ECM consisted of cytarabine (200 mg/m2, 7 days)/mitoxantrone (5 mg/m2, 5 days) combination following 5 days of etoposide (150 mg/m2) which is a standard induction regimen in Japan since the early 1990s [45, 46]. Several studies have compared 100–200 mg/m2 continuous infusion versus high dose (1–3 g/m2/dose infused every 12 h for 3–7 days), but have not demonstrated superiority of high-dose cytarabine [47, 48]. Medical Research Council (MRC) studies in the United Kingdom (the UK) used different schedules of cytarabine in induction: 100 mg/m2/dose intravenous push every 12 h [49]. In MRC AML 9 study, 10 days of cytarabine with daunorubicin/thioguanine was superior to 5-day cytarabine combination, which became the standard induction in the UK. Therefore, cytarabine schedule of either 100–200 mg/m2 continuous infusion or 100 mg/m2/dose intravenous push every 12 h is considered as standard in the contemporary induction regimen.
- (b)
Use of Different Anthracyclines and Their Dose Intensification
There are several formulations of anthracyclines in clinical use: the most common are doxorubicin, daunorubicin, idarubicin (a daunorubicin derivative), and mitoxantrone (an anthracendione derivative). Doxorubicin is not used for AML treatment, because it showed higher mortality from infection and/or gastrointestinal toxicity compared to daunorubicin in the past CALGB study [43]. Among the pediatric studies, AML-BFM 93 compared 3 days of daunorubicin 60 mg/m2 (ADE) and idarubicin 12 mg/m2 (AIE): idarubicin showing faster day 15 blast clearance, but similar EFS and OS rates [50]. MRC AML 12 compared 3 days of daunorubicin 50 mg/m2 (ADE) and mitoxantrone 12 mg/m2 (MAE): better DFS and lower cumulative incidence of relapse were observed in MAE, but CR and OS rates were similar [51]. Recently, liposomal daunorubicin is drawing attention because of low accumulation to heart and high therapeutic index. AML BFM 2004 study compared 3 days of liposomal daunorubicin 80 mg/m2 (ADxE) and idarubicin 12 mg/m2 (AIE): although liposomal daunorubicin was less toxic and more active in t(8;21)-AML, EFS, OS, and relapse incidence were similar between the two arms [52].
Adult studies demonstrated superior CR and OS rates in dose-escalated daunorubicin of 90 mg/m2 for 3 days compared with conventional dose of 45 mg/m2 in combination with cytarabine [53, 54]. However, this approach is not appropriate for children because of increased risk of developing late cardiotoxicity.
In conclusion, the use of either daunorubicin 60 mg/m2 or idarubicin/mitoxantrone 10–12 mg/m2 for 3 days is considered standard in the contemporary induction therapy for children with AML.
- (c)
Addition of Extra Agents to Cytarabine/Anthracycline Backbone
MRC AML 10 trial compared etoposide and thioguanine in combination with cytarabine/daunorubicin induction backbone, but no difference in outcome was observed [55]. Although no clear benefit has been demonstrated for adding extra agents to the standard induction backbone, etoposide is often used in many pediatric AML induction regimens.
One attractive additional agent is gemtuzumab ozogamicin (GO), a calicheamicin conjugated anti-CD33 monoclonal antibody. Because GO demonstrated reasonable activity as a single agent use for relapsed or refractory AML [56, 57], GO combination with standard induction backbone was evaluated in several groups as frontline AML therapy. One adult randomized trial by the Southwest Oncology Group (SWOG) showed no therapeutic efficacies, but higher fatal induction toxicity in GO arm, which led to withdrawal of the product in the US market [58]. However, MRC AML 15 trial in the UK showed benefit of adding GO 3 mg/m2 to one of the three randomized induction regimens for patients with favorable cytogenetics [59], and French ALFA-0701 study showed improved outcome in adult AML patients by adding low fractionated-dose GO (3 mg/m2/dose on days 1, 4, and 7) to 3 + 7 induction [60]. The pediatric study COG AAML0531 demonstrated improved EFS through reduction of relapse rate by adding 3 mg/m2 of GO to the standard regimen [61].
3.3.2.2 Post-Remission Phase
Following remission-induction phase, post-remission therapy is administered aiming to consolidate remission status. It has been clarified in the early adult Eastern Cooperative Oncology Group (ECOG) study in 1983 that all patients eventually relapse without post-remission therapy [62].
- (a)
High-Dose Cytarabine Intensification
The effect of intensifying post-remission chemotherapy was demonstrated by adult CALGB study comparing three different doses of cytarabine in consolidation courses, showing significant improvement in OS for patients <60 years old when a high-dose cytarabine was given, especially those with favorable cytogenetics [63, 64]. In addition, CALGB investigators reported that the outcome of CBF-AML adults who were treated with multiple courses of high-dose cytarabine was better than those who only received one course of high-dose cytarabine [65, 66]. Among the pediatric studies, BFM group showed that high-dose cytarabine with mitoxantrone (HAM regimen) following initial induction was beneficial for t(8;21)-AML [67, 68]. The Japanese pediatric AML trials have incorporated repetitive high-dose cytarabine cycles in consolidation and produced 70% or higher OS rate [46]. Thus, post-remission high-dose cytarabine is considered as standard approach in pediatric AML.
- (b)
Number of Treatment Courses
Optimal number of total treatment courses is an open question. MRC AML 12 trial compared a total of five courses versus four courses of chemotherapy, showing no survival benefit of fifth course of chemotherapy [51]. However, comparison of the two Japanese consecutive trials (AML99 and AML-05) demonstrated conflicting results by different cytogenetic group. While both EFS and OS did not differ in non-CBF-AML children between six and five courses with identical cumulative anthracycline dosage [69], significant worse EFS was observed in CBF-AML children who received five courses of chemotherapy with reduction of cumulative anthracycline dosage [70].
- (c)
Maintenance Therapy
Unlike chemotherapy for ALL, maintenance is unnecessary in the context of contemporary AML therapy with exception of APL. French Leucémie Aiguë Myéloblastique Enfant (LAME) 89 and 91 studies randomized children with AML to receive or not to receive maintenance therapy: strikingly, they not only demonstrated lack of benefit in DFS, but showed inferior OS in the maintenance group [71]. So far, BFM is the only group still incorporating maintenance phase in the frontline pediatric AML therapy [72].
- (d)
Central Nervous System-Directed Therapy
In AML, high leukocyte count, young age (<2 years), monoblastic or myelomonoblastic leukemia (FAB M4 or M5), and cytogenetics of t(8;21), inv(16), or chromosome 11 abnormalities are associated with central nervous system (CNS) leukemia and/or relapse [73]. Outcome of AML patients with CNS involvement is not poor, partly because they are more likely to have favorable cytogenetics thus with lower rate of systemic relapse. Current practice for treatment and prevention of CNS leukemia employs both intrathecal and systemic chemotherapy. Cranial irradiation is not generally used in AML, except the BFM group that continues to administer either 12 or 18Gy of cranial irradiation in AML BFM 2004 trial because of the result from AML BFM 87 trial that observed higher systemic relapse rate in nonirradiated patients [72].
- (e)
Hematopoietic Stem Cell Transplantation for AML
Since the 1980s, allogeneic SCT from matched sibling donor became widely applicable as post-remission therapy for children with AML. Most of the clinical studies conducted between the 1980s to the 1990s allocated patients to allo-SCT by “Mendelian or genetic randomization,” that those with matched sibling donor to allo-SCT and those without to chemotherapy (or autologous SCT). In the Children’s Cancer Group (CCG) 2891 study, allo-SCT had a significantly better DFS and OS than did chemotherapy or auto-SCT [74]. However, no difference was observed in trials AML BFM 98, European Organization of Research and Treatment of Cancer (EORTC) Children’s Leukemia Group (CLG) 58,921, and MRC AML 10 [75]. As risk stratification by cytogenetic abnormalities was introduced from the late 1990s, a consensus was built that no indication of allo-SCT in first CR for favorable cytogenetics such as t(8;21) and inv(16). Along with improved survival by intensive chemotherapy and increasing concern of late effects, allo-SCT is currently restricted to only high-risk cases in first CR.
Since the 2000s, allo-SCTs from alternative donors (e.g., unrelated bone marrow or cord blood donor, haploidentical donor) have been increasingly performed with comparable outcome [76, 77]. Current standard conditioning for pediatric AML is busulfan-based regimen without total body irradiation (TBI), because most of the studies showed identical or even inferior outcome with TBI conditioning for both adult and pediatric AML [78, 79]. In terms of reducing late effects, use of reduced intensity conditioning (RIC) is an attractive option for children undergoing allo-SCT [80–82]. Although there are some promising results, the role of RIC should be carefully evaluated in the context of clinical trial.
3.3.3 Novel Therapeutic Approach for AML
Although approximately 70% of the children with AML are eventually cured, 10% of the patients fail to achieve CR and 30% of the patients suffer from disease recurrence (Table 3.2) [46, 48, 51, 52, 61, 83–86]. One of the well-recognized reinduction regimens for these patients is FLAG, combination of fludarabine, cytarabine, and granulocyte colony-stimulating factor, with or without anthracycline [87]. However, further CR rate is 50–60% and OS rate is 30–40% at most [88]. Improvement in the outcome of frontline treatment also seems to have reached the ceiling with conventional therapeutic approach, since most of the interventions tested in various clinical trials demonstrated negative results or very small improvement. Therefore, development of novel therapeutic approaches is urgently needed to further improve the outcome of AML.
Table 3.2
Results of the recently reported cooperative trials for pediatric AML
Study Group | Trial | Years | No. of patients | Age | %SCT in 1CR | EFS, % (year) | OS, % (year) | RR, % | Reference |
|---|---|---|---|---|---|---|---|---|---|
Japanese childhood AML cooperative study | AML99 | 2000–2002 | 240 | 0–15 | Allo 17% Auto 2% | 61 (5) | 75 (5) | 32 | [46] |
BFM | AML-BFM 2004 | 2004–2010 | 521 | 0–18 | NA | 55 (5) | 74 (5) | 29 | [52] |
JPLSG | AML-05 | 2006–2010 | 443 | 0–18 | 12% | 54 (3) | 73 (3) | 30 | |
St. Jude Children’s Research Hospital | AML02 | 2002–2008 | 216 | 0–21 | 25% | 63 (3) | 71 (3) | 21 | [48] |
AIEOP | AML2002/01 | 2002–2011 | 482 | 0–18 | Allo 29% Auto 21% | 55 (8) | 67 (8) | 24 | [84] |
COG | AAML0531 (GO arm) | 2006–2010 | 1022 | 0–29 | NA | 53 (3) | 69 (3) | 32 | [61] |
NOPHO | AML 2004 | 2004–2009 | 151 | 0–15 | 15% | 57 (3) | 69 (3) | 30 | |
MRC | AML12 | 1995–2002 | 564 | 0–15 | 11% | 54 (10) | 63 (10) | 32 | [51] |
There is substantial number of novel agents under evaluation: new class of chemotherapy (clofarabine, vosaroxin, CPX-351), tyrosine kinase inhibitors (FLT3 inhibitors, KIT inhibitors, RAS pathway inhibitors, Polo-like kinase inhibitor, Aurora-kinase inhibitor), proteasome inhibitors (bortezomib, carfilzomib), epigenetic agents (methyltransferase inhibitors, histone deacetylases, DOT1L inhibitor), immunotherapy [SGN33A, bispecific T-cell engager (BiTE) antibodies, chimeric antigen receptor (CAR) T cells], etc. [89]. It is expected that the genomic landscape of AML would be fully unmasked in the near future and that the integration of discovered genomics into contemporary therapy would progress in order to realize precision medicine which would result in further improvement in the outcome of childhood AML.
3.4 Myeloid Leukemia Associated with Down Syndrome
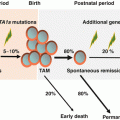
Down syndrome (DS) results from trisomy 21 and occurs in 1 in 700–1000 births [90, 91]. Although the incidence of neoplasms in DS does not differ significantly from that in the general population, the distribution of malignancies is different [92]. Patients with DS show a unique spectrum of malignancies, with a 10- to 20-fold higher risk of acute leukemia and a lower incidence of solid tumors [92, 93]. The most frequent form of leukemia during childhood, both with and without DS, is ALL, and the incidence of ALL in children with DS is approximately 20-fold higher than that in non-DS children [92]. However, the most marked increase in incidence in DS infants is AMKL, known as “myeloid leukemia associated with DS (ML-DS).” The development of ML-DS is closely linked to a preceding temporary form of neonatal leukemia called transient abnormal myelopoiesis (TAM). TAM and ML-DS are classified as an independent category as myeloid proliferations of Down syndrome in recent WHO classification [2, 94, 95]. Both generally manifest as megakaryoblastic phenotype, with TAM occurring at birth or within days of birth and being resolved in 1–2 months. About 1–2% of children with DS develop AML during the first 4 years of life. MDS often precedes AMKL and can last for several months. The clinical course of patients of MDS in DS appears to be relatively indolent, presents initially with a period of thrombocytopenia, and lacks significant increase of blasts. Findings from recent clinical and laboratory studies revealed that quantitative alterations of RUNX1, ETS2, and ERG genes (all located at 4-Mb region of chromosome 21) that resulted from constitutional trisomy 21 up-regulate GATA1 mutations and cause TAM [96] and that additional genetic alterations including those in epigenetic regulators and signaling molecules are involved in the progression from TAM to ML-DS [97]. Children with DS above the age of 5 with AML or MDS have different biology to ML-DS, such as non-megakaryoblastic phenotype and no GATA1 mutation of blasts, and should be considered not as “typical ML-DS,” but as “conventional” AML or MDS.
ML-DS has unique characteristics of higher sensitivity to chemotherapeutic agents. In vitro studies showed that ML-DS blasts were significantly more sensitive to chemotherapeutic drugs than non-DS AML cells [98]. ML-DS blasts are especially sensitive to cytarabine, possibly because of the effect of GATA1 mutations and trisomy 21 on the levels of cytarabine-metabolizing enzymes [99]. Before the 1990s, most patients with ML-DS received suboptimal therapy, resulting in poor outcomes. In 1992, high rates of EFS with intensive AML treatment were reported from the Pediatric Oncology Group (POG) [100]. After recognition of the favorable outcome of ML-DS patients treated with the AML protocol, recruitment to collaborative studies for ML-DS patients increased, but it became apparent that treatment-related toxicity was high in most series [101–103]. Since then, several collaborative groups have adapted their AML protocols for ML-DS by reducing the dosage of chemotherapeutic agents [103]. As a result, in recent clinical studies in several countries, ML-DS children were treated separately and less intensively than non-DS AML children. Recent clinical trials for ML-DS are summarized in Table 3.3 [101–109]. In Europe, the ML-DS 2006 study by the I-BFM-SG, which was based on the reduced intensity arm of the BFM 98 protocol with further elimination of etoposide in consolidation courses, was conducted. This protocol consists of four courses including high-dose cytarabine. The US AAML0431 study, conducted from 2007 to 2011 by the COG, which also included high-dose cytarabine, showed excellent outcomes [110]. The Japanese trials for ML-DS had investigated less intensive chemotherapy compared with those conducted in Western countries, and treatment outcomes were comparable [108, 109, 111]. Toronto group reported long-term results of an ultralow dose cytarabine-based regimens suggested that at least some population of ML-DS could be cured by minimum intensive therapy [106]. Predicting prognostic factors had been examined by several studies; however, no universal factors had been found to date [107, 109, 112]. On the other hand, it is reported that relapsed/refractory cases are rarely salvageable, with very limited role of allo-SCT [113]. In terms of treatment outcome, ML-DS is a heterogeneous disease and risk-oriented therapy is a reasonable strategy, so that finding an accurate method, perhaps MRD, for identifying a subgroup with poor prognosis is urgently needed. Moreover, new therapeutic approaches for relapsed/refractory cases using new drugs such as Wee 1 inhibitor [114], Aurora-kinase inhibitor [115], and histone deacetylase inhibitors [116] will be also needed.
Table 3.3
 Inherited Bone Marrow Failure Syndrome, TAM
Inherited Bone Marrow Failure Syndrome, TAM
 Childhood Aplastic Anemia
Childhood Aplastic Anemia
 Hemophagocytic Lymphohistiocytosis, Secondary
Hemophagocytic Lymphohistiocytosis, Secondary
 Primary Hemophagocytic Lymphohistiocytosis
Primary Hemophagocytic Lymphohistiocytosis
 Myelodysplastic Syndrome (MDS) and Juvenile Myelomonocytic Leukemia (JMML)
Myelodysplastic Syndrome (MDS) and Juvenile Myelomonocytic Leukemia (JMML)
 Hematopoietic Stem Cells: The Basis of Normal and Malignant Hematopoiesis
Hematopoietic Stem Cells: The Basis of Normal and Malignant Hematopoiesis




Outcome of recent clinical trials for myeloid leukemia with Down syndrome
Related posts:
Inherited Bone Marrow Failure Syndrome, TAM
Childhood Aplastic Anemia
Hemophagocytic Lymphohistiocytosis, Secondary
Primary Hemophagocytic Lymphohistiocytosis
Myelodysplastic Syndrome (MDS) and Juvenile Myelomonocytic Leukemia (JMML)
Hematopoietic Stem Cells: The Basis of Normal and Malignant Hematopoiesis
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
