Abstract
Acute lymphoblastic leukemia (ALL) is the most common malignancy of childhood with a peak incidence between 2 and 6 years of age. Recent advances in understanding the leukemia biology, refinement in risk stratification and large international cooperative group clinical trials have led to a remarkable improvement in the outcome of pediatric ALL. Overall, current treatment regimens can cure more than 80% of patients. We are now able to identify a subset of patients who can be effectively be cured with less intensive therapy; however, about 20% of patients will ultimately relapse, making relapsed leukemia one of the leading causes of cancer-related deaths. Efforts are underway to identify novel strategies to appropriately identify these patients who are at high risk of relapse and to optimize therapy for them. This chapter provides a brief overview of ALL risk factors, clinical features, diagnostic modalities, prognostic factors, and current treatment schema for various risk groups of newly diagnosed and relapsed ALL.
Keywords
Childhood leukemia, risk stratification, minimal residual disease assessment, leukemia chemotherapy, leukemia targeted therapy
Acute leukemias represent a clonal expansion and arrest at a specific stage of normal myeloid or lymphoid hematopoiesis. They constitute 97% of all childhood leukemias and consist of the following types:
- •
Acute lymphoblastic leukemia (ALL)—75%: Amongst ALL, the nomenclature of the subtypes has been changed from “precursor B-lymphoblastic leukemia/lymphoma” and “precursor T-lymphoblastic leukemia/lymphoma” to “B or T-lymphoblastic leukemia/lymphoma” respectively.
- •
Acute myeloblastic leukemia (AML), also known as acute non-lymphocytic leukemia—20%.
- •
Acute undifferentiated leukemia—<0.5%.
- •
Acute mixed-lineage leukemia.
Chronic myeloid leukemias constitute 3% of all childhood leukemias and consist of
- •
Philadelphia chromosome-positive (Ph 1 positive) myeloid leukemia.
- •
Juvenile myelomonocytic leukemia.
Etiology
The etiology of acute leukemia is unknown. The following factors are important in the pathogenesis of leukemia:
- •
Ionizing radiation.
- •
Chemicals (e.g., benzene in AML)
- •
Drugs (e.g., use of alkylating agents either alone or in combination with radiation therapy increases the risk of AML).
- •
Genetic considerations:
- •
Identical twins—If one twin develops leukemia during the first 5 years of life the risk of the second twin developing leukemia is 20%.
- •
Incidence of leukemia in siblings of leukemia patient is four times greater than that of the general population.
- •
Group
Risk
Time interval
Trisomy 21 (Down syndrome)
1 in 95
<10 years of age
Bloom syndrome
1 in 8
<30 years of age
Fanconi anemia
1 in 12
<16 years of age
- •
Increased incidence with the following genetically determined conditions:
- •
Congenital agammaglobulinemia.
- •
Poland syndrome.
- •
Shwachman–Diamond syndrome.
- •
Ataxia telangiectasia.
- •
Li–Fraumeni syndrome (germline p53 mutation)—the familial syndrome of multiple cancers in which acute leukemia is a component malignancy.
- •
Neurofibromatosis.
- •
Diamond–Blackfan anemia.
- •
Kostmann disease.
- •
Bloom syndrome.
- •
- •
Most cases of leukemia do not stem from an inherited genetic predisposition but from somatic genetic alterations. However, recent studies indicate a possible genetic linkage with inherited polymorphisms in ARID5B and IKZF1 genes for childhood ALL.
Clinical Features of ALL
Table 18.1 shows the common clinical and laboratory presenting features in ALL.
| Clinical and laboratory presenting features | Percentage of patients |
|---|---|
| SYMPTOMS AND PHYSICAL FINDINGS | |
| Fever | 61 |
| Bleeding (e.g., petechia or purpura) | 48 |
| Bone pain | 23 |
| Lymphadenopathy | 50 |
| Splenomegaly | 63 |
| Hepatosplenomegaly | 68 |
| LABORATORY FEATURES | |
| Leukocyte count (mm 3 ) | |
| <10,000 | 53 |
| 10,000–49,000 | 30 |
| >50,000 | 17 |
| Hemoglobin (g/dl) | |
| <7.0 | 43 |
| 7.0–11.0 | 45 |
| >11.0 | 12 |
| Platelet count (mm 3 ) | |
| <20,000 | 28 |
| 20,000–99,000 | 47 |
| >100,000 | 25 |
| Lymphoblast morphology | |
| L1 | 84 |
| L2 | 15 |
| L3 | 1 |
General Systemic Effects
- 1.
Fever (60%).
- 2.
Lassitude (50%).
- 3.
Pallor (40%).
Hematologic Effects Arising from Bone Marrow Invasion
- 1.
Anemia—causing pallor, fatigability, tachycardia, dyspnea, and sometimes congestive heart failure.
- 2.
Neutropenia—causing fever, ulceration of buccal mucosa, and infection.
- 3.
Thrombocytopenia—causing petechia, purpura, easy bruisability, bleeding from mucous membrane, and sometimes internal bleeding (e.g., intracranial hemorrhage).
- 4.
One to 2% of patients present initially with pancytopenia and may be erroneously diagnosed as having aplastic anemia or bone marrow failure (represents 5% of acquired aplastic anemia) and ultimately develop acute leukemia. In these cases the illness is characterized by:
- •
Pancytopenia or single cytopenia.
- •
Hypocellular bone marrow.
- •
No hepatosplenomegaly.
- •
Diagnosis of leukemia 1–9 months after onset of symptoms.
- •
The treatment consists of supportive transfusions initially and specific antileukemic chemotherapy, when leukemia is diagnosed.
Clinical Manifestations Arising from Lymphoid System Infiltration
- 1.
Lymphadenopathy—sometimes presents with bulky mediastinal lymphadenopathy causing superior vena cava syndrome. This is more common in T-cell leukemia in adolescents.
- 2.
Splenomegaly.
- 3.
Hepatomegaly.
Clinical Manifestations of Extramedullary Invasion
Central Nervous System Involvement
This occurs in less than 5% of children with ALL at initial diagnosis. It may present with the following:
- •
Signs and symptoms of raised intracranial pressure (e.g., headache, morning vomiting, papilledema, bilateral sixth-nerve palsy).
- •
Signs and symptoms of parenchymal involvement (e.g., focal neurologic signs such as hemiparesis, cranial nerve palsies, convulsions, cerebellar involvement—ataxia, dysmetria, hypotonia, hyperflexia).
- •
Hypothalamic syndrome (polyphagia with excessive weight gain, hirsutism, and behavioral disturbances).
- •
Diabetes insipidus (posterior pituitary involvement).
- •
Chloromas of the spinal cord (very infrequent in ALL)—may present with back pain, leg pain, numbness, weakness, Brown–Séquard syndrome, and bladder and bowel sphincter problems.
- •
Central nervous system (CNS) hemorrhage—complication that occurs more frequently in patients with AML than in ALL. It is caused by:
- •
Leukostasis in cerebral blood vessels, leading to leukothrombi, infarcts, and hemorrhage.
- •
Thrombocytopenia and coagulopathy, contributing to CNS hemorrhage.
- •
Genitourinary Tract Involvement
Testicular Involvement
- 1.
Usually presents with painless enlargement of the testis.
- 2.
Occurs in less than 2% of boys at diagnosis. Isolated testicular relapse accounts for less than 10% of all ALL relapses.
Ovarian Involvement
Occurs very rarely.
Priapism
Occurs rarely. It is due to involvement of sacral nerve roots or mechanical obstruction of the corpora cavernosa and dorsal veins by leukemic infiltrates.
Renal Involvement
- 1.
Occasionally may present with hematuria, hypertension, and renal failure.
- 2.
Evaluated by ultrasonography; renal involvement is more common in T-cell ALL or mature B-cell ALL (Burkitt).
Gastrointestinal Involvement
- 1.
The gastrointestinal (GI) tract may be involved in ALL. The most common manifestation is bleeding.
- 2.
Leukemic infiltrates in the GI tract are usually clinically silent until terminal stages when necrotizing enteropathy might occur. The most common site for this is the cecum, giving rise to a syndrome known as typhlitis.
Bone and Joint Involvement
Bone pain is one of the initial symptoms in 25% of patients. It may result from direct leukemic infiltration of the periosteum, bone infarction, or expansion of marrow cavity by leukemic cells. Radiologic changes occasionally seen include:
- •
Osteolytic lesions involving medullary cavity and cortex.
- •
Transverse metaphyseal radiolucent bands.
- •
Transverse metaphyseal lines of increased density (growth arrest lines).
- •
Subperiosteal new bone formation.
Skin Involvement
Skin involvement occurs occasionally in neonatal leukemia or AML.
Cardiac Involvement
One-half to two-thirds of patients have demonstrated cardiac involvement at autopsy, although symptomatic heart disease occurs in less than 5% of cases due to late toxicity of treatment. Pathologic findings may include leukemic infiltrates and hemorrhage of the myocardium or the pericardium.
Lung Involvement
Lung involvement is uncommon and may be due to leukemic infiltrates or hemorrhage in patients with very high white blood cell (WBC) counts (leukostasis).
Diagnosis
Laboratory Studies
- 1.
Blood count
- a.
Hemoglobin : Moderate to marked reduction. Normocytic; normochromic red cell morphology. Low hemoglobin indicates longer duration of leukemia; higher hemoglobin may indicate a more rapidly proliferating leukemia.
- b.
WBC count : Low, normal, or increased.
- c.
Blood smear : Blasts are present on blood smear. Very few to none (in patients with leukopenia). When the WBC count is greater than 10,000/mm 3 , blasts are usually abundant. Eosinophilia is seen uncommonly in children with ALL.
- d.
Thrombocytopenia : 92% of patients have platelet counts below normal. Serious hemorrhage (GI or intracranial) occurs at platelet counts less than 20,000/mm 3 .
- a.
- 2.
Bone marrow : Bone marrow is usually replaced by 80–100% blasts. Megakaryocytes are usually absent. Leukemia must be suspected when the bone marrow contains more than 5% blasts. The hallmark of the diagnosis of acute leukemia is the blast cell, a relatively undifferentiated cell with diffusely distributed nuclear chromatin, one or more nucleoli (more prominent in AML) and scant cytoplasm (more abundant in AML). Special bone marrow studies, which help in detailed cell classification, include the following:
- a.
Immunophenotyping using flow cytometry
- b.
Cytogenetics—karyotype and molecular studies.
- a.
- 3.
Chest radiograph : Mediastinal mass in T-cell leukemia.
- 4.
Blood chemistry : Electrolytes, blood urea, uric acid, LDH, liver function tests, immunoglobulin levels.
- 5.
Cerebrospinal fluid (CSF): Chemistry, cell count, and cytospin for pathological examination. Cerebrospinal fluid findings for the diagnosis of CNS leukemia require:
- a.
Presence of >5 WBCs/mm 3 .
- b.
Identification of blast cells on cytocentrifuge examination. CNS involvement in leukemia is classified as follows:
- i.
CNS1 <5 WBCs/mm 3 , no blasts on cytocentrifuge slide.
- ii.
CNS2 <5 WBCs/mm 3 , blasts on cytocentrifuge slide.
- iii.
CNS3 >5 WBCs/mm 3 , blasts on cytocentrifuge slide.
- i.
- c.
TdT stain for suspicious cells.
- d.
If a lumbar puncture is traumatic in a patient with peripheral blasts the following formula can be helpful in defining the presence of CNS leukemia. CNS disease is present if:
- i.
CSF WBC/CSF RBC>2×Blood WBC/Blood RBC.
- i.
- a.
- 6.
Coagulation profile : PT, PTT, and fibrinogen.
- 7.
Cardiac function : Electrocardiogram and echocardiogram.
- 8.
Infectious disease profile : Varicella antibody titer, cytomegalovirus antibody titer, herpes simplex antibody, hepatitis antibody screening.
- 9.
Immunologic screening : Serum for immunoglobulin levels, C3 and C4.
Classification
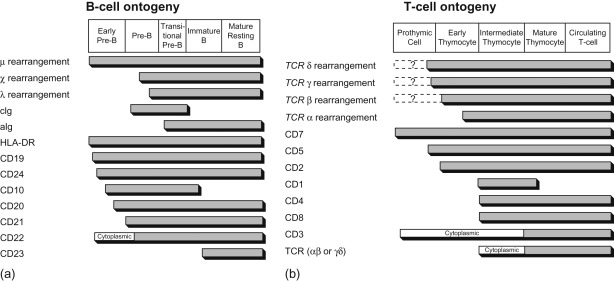
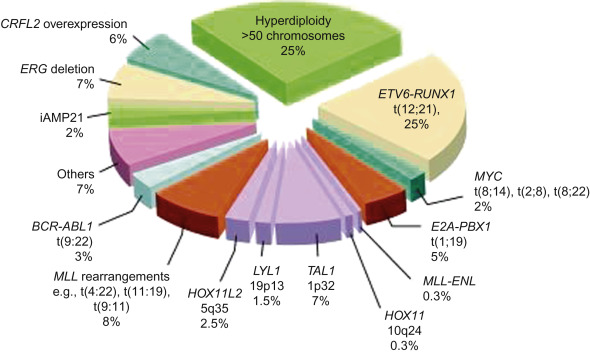
Acute leukemia can be classified based on morphologic characteristics ( Table 18.2 ), cytochemical features ( Table 18.3 ), immunologic characteristics ( Figure 18.1 ), and cytogenetic and molecular characteristics ( Figure 18.2 ). The World Health Organization has developed a new classification of ALL based on cytogenetic and molecular characteristics ( Table 18.4 ).
| Cytologic features a | L1 | L2 | L3 b |
|---|---|---|---|
| Cell size | Small cells predominate | Large, heterogeneous in size | Large and heterogeneous |
| Nuclear chromatin | Homogeneous | Variable, heterogeneous | Finely stippled and homogeneous |
| Nuclear shape | Regular, occasional clefting or indentation | Irregular, clefting and indentation common | Regular, oval to round |
| Nucleoli | Not visible, or small and inconspicuous | One or more present, often large | Prominent, one or more vesicular |
| Amount of cytoplasm | Scanty | Variable, often moderately abundant | Moderately abundant |
| Basophilia of cytoplasm | Slight or moderate, rarely intense | Variable, deep in some | Very deep |
| Cytoplasmic vacuolation | Variable | Variable | Often prominent |
a For each of the features considered, up to 10% of the cells may depart from the characteristic of the type.
b The only immunologically pure type of ALL that can be consistently recognized morphologically and invariably carries IgM surface receptor on its membrane.
| Staining reaction | ALL | AML | Acute nonlymphoblastic leukemia | ||
|---|---|---|---|---|---|
| Acute myelomonocytic leukemia | Erythroleukemia | Megakaryoblastic leukemia | |||
| NONENZYMATIC | |||||
| PAS | Present as coarse granules or blocks in a variable number of cells | Negative or diffusely positive | Negative or fine granulation | Strongly positive granular | Positive or negative |
| Sudan black | Negative | Positive | Positive | Positive | Negative |
| ENZYMATIC | |||||
| Peroxidase | Negative | Positive a | Usually negative | Positive | Negative |
| Alkaline phosphatase | Normal | Low | High | Normal or high | – |
| ESTERASES | |||||
| Naphthol ASD Chloroacetate | Negative | Positive | Negative | Negative | |
| Naphthol ASD acetate | Negative or weakly positive | Positive (not inhibited by fluoride) | Strongly positive (inhibited by fluoride) | Weakly positive | Positive or negative |
| α-Naphthyl acetate | Negative | Negative | Strongly positive | Strongly positive | Positive or negative |
| Acid phosphatase | Positive in T-ALL | Negative | Negative | Negative | Positive (localized pattern) |
a The peroxidase reaction, when positive, is considered to indicate the presence of myeloblastic rather than lymphoblastic elements. Unfortunately, myeloblasts that contain no specific granules will be peroxidase-negative. It is these cells that cause the greatest difficulty in morphologic classification. Generally, when morphologic classification is difficult, these histochemical tests are of little help. However, demonstration of MPO (myeloperoxidase) by immunologic techniques or expression of MPO by molecular methods can be performed in specialized research laboratories.
|
Light microscopy, cytochemistry, immunophenotyping, and cytogenetics are necessary studies to characterize leukemic subtypes. Current risk classification schema and treatment regimens however primarily utilize immunophenotypic and cytogenetic features only.
Morphology
Light Microscopy
Cytochemical criteria have been established to differentiate lymphoblasts from myeloblasts ( Table 18.3 ). ALL can be further subclassified according to the French–American–British classification as L1, L2, and L3 morphologic types ( Table 18.2 ).
Immunology
The putative immunologic classification and cellular characteristics of B-lineage ALL and T-cell ALL are shown in Figure 18.1 .
A panel of antibodies is used to establish the diagnosis of leukemia and to distinguish among the immunologic subclones. The panel should include at least one marker that is highly lineage-specific, for example, CD19 for B-lineage, cytoplasmic CD3 for T lineage and myeloperoxidase or markers of monocytic differentiation such as non-specific esterase, CD11c, CD14, CD64, lysozyme for myeloid lineage malignancies. In addition, the use of cytoplasmic CD79a, cytoplasmic CD22, CD10 for B-lineage, surface CD3, CD7, and CD5 for T lineage and CD13 and CD33 for myeloid cells can be helpful in differentiating unclear immunophenotypes.
Immunophenotype Distribution of ALL
B-precursor cell accounts for 80% of ALL cases.
T-cell accounts for 15–20% of ALL cases. This subtype is associated with
- •
Older age at presentation.
- •
High initial white cell count.
- •
Presence of extramedullary disease, for example, mediastinal mass.
- •
Traditionally poor prognosis but treatment on high-risk intensive therapies has improved the outcome.
Mature B-cell accounts for 1–2% of ALL cases. These are surface immunoglobulin-positive and are treated as Burkitt’s lymphoma. The prognosis is similar to other subtypes of high-risk ALL.
Acute Leukemia of Ambiguous Lineage
Please see discussion in Chapter 19 .
Cytogenetics and Molecular Characteristics
The cytogenetic abnormalities observed in leukemia have biologic and prognostic significance. The realization of the biologic significance of leukemia cytogenetics has resulted in the appreciation of distinct biological subsets associated with prognosis ( Table 18.5 ) and treatment stratification.
| Chromosomal abnormalities | EFS |
|---|---|
| Trisomies of chromosomes 4, 10, and 17 | 89% 8 years |
| ETV6-RUNX1 Fusion positive | 86% 5 years |
| Hypodiploidy <45 chromosomes | 39% 8 years |
| MLL-REARRANGED | |
| Infants (0–12 months) | 37% 4 years |
| Non infants (more than 12 months) | 50% 8 years |
| t(9;22) | 39% 8 years a |
a Studies have shown a 3-year EFS of 80% using intensive chemotherapy with Imatinib.
Molecular Genetics of ALL
Figure 18.2 shows the distribution of molecular rearrangements in childhood ALL.
Seventy-five percent of childhood ALL cases have evidence of chromosomal gains/losses and/or translocations:
- •
Hyperdiploid ALL is characterized by whole chromosomal gains and is observed in 30% of patients. The gains are non-random (trisomy 21 is the most common). Specific trisomies (4, 10 and 17 and 18 in some studies) are associated with a particularly good outcome.
- •
Hypodiploid ALL is found in approximately 6% of patients, characterized by fewer than 46 chromosomes in the leukemic blasts. Patients with either a karyotype with <44 chromosomes or a DNA index of <0.81 have a worse outcome.
- •
ETV6-RUNX1 fusion gene t(12;21) (p13q22). t(12;21), previously referred to as TEL-AML1 is detected by standard cytogenetics in less than one in 1000 cases; whereas, using molecular techniques such as fluorescent in situ hybridization (FISH), it is detected in approximately 25% of B-ALL cases. This translocation is associated with an excellent prognosis.
- •
BCR–ABL fusion gene t(9;22) (q34q11). t(9;22) occurs in only 3% of pediatric ALL cases. This is in contrast to adult ALL where this translocation is present in 25% of cases and 95% of chronic myelogenous leukemia (CML) patients. In pediatric BCR–ABL-positive ALL, the BCR breakpoint characteristically produces a 190-kDa protein (p190) in contrast to CML where a different protein (p210) is usually produced. The t(9;22) in pediatric ALL is usually associated with older age, higher WBC count, and frequent CNS involvement at diagnosis.
- •
TCF3-PBX1 fusion gene t(1;19) (q23p13.3). t(1;19) is frequently associated with an elevated WBC at diagnosis and occurs in approximately 5% of newly diagnosed ALL. While earlier studies have indicated it as a relatively poor prognostic marker, the contemporary intensive treatment on current ALL regimens has shown no difference in the outcomes.
- •
On the other hand, TCF3-HLF fusion gene t(17:19) occurs in approximately 1% of newly diagnosed patients and has been associated with hypercalcemia, DIC and poor prognosis. This translocation will be classified under “very high category” in the upcoming COG classification schema, and patients will receive intensive upfront treatment including the option of stem cell transplant in first remission.
- •
MLL gene rearrangement at chromosome band 11q23 affects 80% of ALL cases in infants, 3% of ALL cases in older children, and 85% of secondary AML involving the use of topoisomerase II inhibitors. This translocation carries a very poor prognosis despite intensive therapy.
- •
Intrachromosomal amplification of chromosome 21 (iAMP21), being defined as at least four copies of RUNX1 in a single chromosome, is found in approximately 2% of pediatric B-ALL patients. iAMP21 is associated with inferior outcome, particularly for standard-risk patients, however, outcomes are better when these patients are treated with intensive chemotherapy regimens.
- •
Mature B-cell ALL translocations involve MYC genes on chromosome 8q24. Eighty percent of mature B-ALL cases contain t(8;14) (q24q32); the remaining cases have t(2;8) (p12q24) or t(8;22) (q24q11). All of these translocations deregulate MYC expression through association with highly active immunoglobulin genes and need to be treated with short and intensive chemotherapy comprising the same agents used to treat Burkitt lymphoma.
- •
More than 50% of the cases of T-cell ALL have activating mutations that involve NOTCH1 , a gene encoding a transmembrane receptor that regulates normal T-cell development. An additional 20% of cases have mutations in FBXW7 which degrades intracellular NOTCH. When NOTCH receptors become activated there is a cascade of proteolytic cleavages, which causes intracellular NOTCH1 to translocate into the nucleus, which then regulates the transcription of a set of oncogenes including MYC . It is believed that aberrant NOTCH signaling causes constitutive expression of MYC and activation of RAS . Interference with NOTCH signaling by small-molecule inhibition has the potential for induction of remission in T-cell ALL.
Future Directions in ALL Classifications
- 1.
Gene expression profiling using DNA microarray technology and next-generation sequencing technology can define biologically and prognostically distinctive ALL subsets, identify genes which may be responsible for leukemogenesis and may identify genes for which targeted therapy could be developed. The discovery of “Ph-like” ALL in up to 15% of patients (older age, inferior prognosis) that is associated with novel tyrosine kinase fusions and JAK mutations, provides an opportunity for targeted therapy with tyrosine kinase and JAK inhibitors.
- 2.
Host pharmacogenomics: Polymorphisms for genes that encode drug-metabolizing enzymes can influence the efficacy and toxicity of chemotherapy. Gene polymorphisms for thiopurine methyltransferase is a gene that catalyzes the inactivation of mercaptopurine. Ten percent of the population carries at least one variant allele. This results in high levels of active metabolites of mercaptopurine. These patients, especially homozygous, have an increased risk of side effects and require marked reduction of 6-mercaptopurine doses.
- 3.
Identification of high-risk genetic lesions in newly diagnosed ALL: Investigators have identified DNA copy number abnormalities in genes that encode regulators of B-cell development, such as CDKN2A/B , PAX5 , IKZF1 , and EBF1 . The alteration of IKZF1 , a gene that encodes the lymphoid transcription factor IKAROS , has been associated with poor outcome. A deletion or a mutation of IKZF1 is strongly associated with elevated levels of MRD. In addition, JAK2 mutations, somatic alterations in RAS pathway genes ( NRAS, KRAS, PTPN11 , and NF1 ) and CRLF2 alterations have been identified in increased frequency in high-risk patients. The Janus genes JAK1 and JAK2 have been shown to be mutated in Down syndrome patients with B-lymphoblastic ALL and T-lymphoblastic ALL as well. Current high-risk Children’s Oncology Group (COG) ALL trials are utilizing these markers to determine their frequency and prognostic significance, with a goal to further refine the risk stratification schema.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


