The acute leukemias of childhood are rare diseases that collectively represent about 30% of malignancies in children younger than age 15 years. Approximately 3000 new cases of childhood leukemia occur annually in the United States, 80% of which are acute lymphoblastic leukemia (ALL). Between the 1960s and 1990s, the prognosis for children with ALL improved dramatically. With current multiagent chemotherapy regimens, 80% to 90% of children with ALL are long-term relapse-free survivors. In this chapter, childhood ALL is reviewed with respect to classification, molecular pathogenesis, clinical presentation, laboratory findings, differential diagnosis, treatment strategies, prognosis, and late effects of therapy. Causative and epidemiologic considerations, including the incidence, prevalence, and life span of afflicted individuals, are discussed in Chapter 40 . Discussion of the cytogenetic aspects of the leukemias appears in Chapter 45 , and the molecular basis of human malignancy is further discussed in Chapter 33 . Details concerning the various chemotherapeutic agents are provided in Chapter 46 , and myelogenous leukemias and myeloproliferative disorders are discussed in Chapter 51 .
Classification
The childhood leukemias can be classified as acute or chronic. Acute leukemia is characterized by clonal expansion of immature hematopoietic or lymphoid precursors, whereas chronic leukemia refers to conditions characterized by the expansion of mature marrow elements. Congenital leukemia refers to leukemias diagnosed within the first 4 weeks of life.
Acute leukemia is characterized by replacement of normal marrow elements with malignant blast cells, relatively undifferentiated cells with diffusely distributed nuclear chromatin, one or more nucleoli, and basophilic cytoplasm. A number of methods exist for characterizing malignant blast cells, including morphology, cytochemistry, and immunophenotype, as well as chromosomal and molecular genetic aberrations. Approximately 80% of cases of childhood acute leukemia are lymphoblastic.
Morphologic and Cytochemical Classification
The production of blast forms is part of the normal maturational sequence of hematopoietic and lymphoid elements. Blast cells are primitive precursors, lacking many of the features of differentiation. Under normal conditions, blast forms constitute fewer than 5% of the nucleated cells of the bone marrow. Blast cells are usually not observed in the peripheral blood, except during periods of profound overproduction of blood cells in response to infection or bleeding or from bone marrow invasion by granulomas, fibrosis, or tumor cells (leukoerythroblastosis). Leukemic blast cells can be difficult to distinguish morphologically from normal, nonmalignant blasts, although the finding of more than 5% blast forms in the marrow, or the presence of blast cells in the peripheral blood, should raise the suspicion of leukemia. In acute leukemia, patients often present with a marrow that is almost fully replaced by blast forms.
Once the diagnosis of leukemia is made, it is sometimes difficult to differentiate a lymphoid from a myeloid blast. The morphologic characteristics of lymphoblasts are shown in Figure 50-1 . Wright-Giemsa–stained lymphoblasts have smooth, homogeneous nuclear material with indistinct nucleoli and only a small rim of light blue–staining cytoplasm, generally without granules. In contrast, myeloblasts differ from lymphoblasts in that the former have a lower nuclear-to-cytoplasmic ratio, more finely developed nuclear chromatin, and more distinct punched-out nucleoli. Cytoplasmic granules are often present in myeloblasts, and the detection of eosinophilic Auer rods is pathognomonic. However, small myeloblasts may be confused with lymphoblasts morphologically, although they can usually be distinguished by flow cytometric or cytochemical studies.
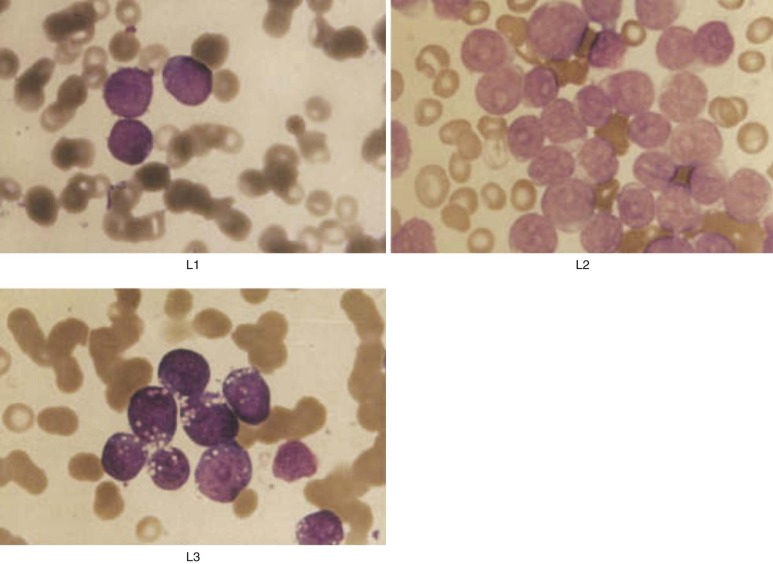
A standardized morphologic classification system devised by the French-American-British (FAB) Cooperative Working Group is generally used to categorize the appearance of leukemic blasts. In this system, ALL is subdivided into three morphologic categories: L1, L2, and L3 (see Fig. 50-1 ). L1 is the most common subtype, observed in about 90% of cases of childhood ALL. L1 lymphoblasts are small cells characterized by a high nuclear-to-cytoplasmic ratio. The pale blue cytoplasm is scanty and limited to a small portion of the perimeter of the cell. The cells have indistinct nucleoli and nuclear membranes that vary from round to clefted. Cells in the L2 category, found in 5% to 15% of pediatric cases, are larger than those classified as L1, show marked variability in size, and have prominent nucleoli and more abundant cytoplasm. They may be difficult to distinguish morphologically from the M1 variant of myeloid leukemia, and thus flow cytometric analysis of cell surface markers is essential to differentiate ALL from acute myelogenous leukemia (AML). L1 and L2 lymphoblasts do not differ significantly in terms of cell surface markers, genetic abnormalities, or prognosis. Only 1% to 2% of ALL pediatric patients have L3 lymphoblasts, which appear identical to Burkitt lymphoma cells, with deeply basophilic cytoplasm and prominent cytoplasmic vacuolization. L3 morphology is almost always associated with a mature B-cell phenotype (including expression of surface immunoglobulin) and translocations involving the MYC gene on chromosome 8; patients with these characteristics should be treated with regimens for advanced-stage Burkitt lymphoma rather than ALL. Other morphologic variants of ALL, including ALL with hand mirror cells or granules, have been described but do not appear to have prognostic significance.
Various cytochemical stains can distinguish ALL from AML. In approximately 80% of cases of ALL, lymphoblasts react positively with periodic acid–Schiff (PAS), which stains cytoplasmic glycogen. Myeloid-specific stains, such as myeloperoxidase, specific and nonspecific esterase, and Sudan black (a cytoplasmic lipid stain), are usually negative in ALL. In contrast, myeloblasts are myeloperoxidase positive in 75% of patients. Terminal deoxynucleotidyl transferase (TdT), an enzyme that catalyzes the polymerization of deoxynucleoside monophosphates into a single-strand DNA primer, can be demonstrated in most cases of ALL but is rarely present in cases of myeloid leukemia.
Immunophenotype
ALL is subcategorized based on the expression of lineage- and maturation-specific antigens present on the cell surface and in the cytoplasm of lymphoblasts. In addition, immunophenotypic studies of lymphoblasts have provided important insights into the differentiation and maturation of normal B and T lymphocytes. Because the leukemic blast represents the neoplastic expansion of a lymphoid progenitor cell, many phenotypic and genotypic features of the lymphoblast mimic those of the normal lymphoid cell counterpart of specific lineage and maturational stage. Figure 50-2 depicts a schematic overview of lymphoid cell differentiation.
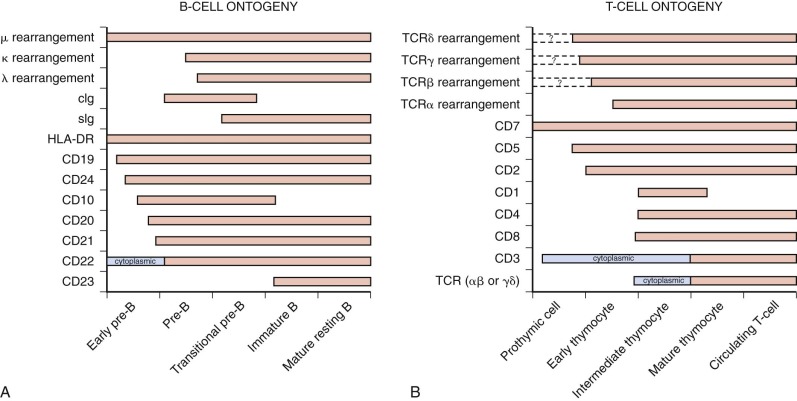
Based on reactivity with a panel of lineage-associated antibodies, ALL has been subclassified into three broad categories: B-precursor cell, mature B cell, and T cell. The immunophenotypic subsets are associated with distinctive clinical features ( Table 50-1 ).
| Parameter | Early Pro–B Cell, CD10− | Early B-Precursor Cell, CD10+ | Pre–B Cell (cIgM+) | T Cell |
|---|---|---|---|---|
| No. of patients | 52 | 635 | 156 | 124 |
| Gender (% male) | 39% | 53% | 50% | 75% |
| Age (yr) | ||||
| <1 | 33% | 1% | 6% | 1% |
| 1 to <10 | 50% | 82% | 80% | 62% |
| ≥10 | 17% | 17% | 14% | 37% |
| Leukocyte count (× 10 9 /L) | ||||
| ≤20 | 38% | 75% | 53% | 23% |
| >50 | 44% | 11% | 21% | 57% |
| Platelet count ≤100 × 10 9 /L | 77% | 75% | 81% | 56% |
| Hemoglobin ≤8 g/dL | 58% | 40% | 60% | 15% |
| Splenomegaly * | 50% | 34% | 46% | 57% |
| Hepatomegaly * | 56% | 46% | 48% | 61% |
| Mediastinal mass | 0% | 0% | 1% | 72% |
| Lymphadenopathy | 35% | 36% | 41% | 78% |
| CNS-positive (CNS-3) † | 10% | 1% | 1% | 11% |
* >4 cm below the costal margin.
† CNS-3, diagnostic cerebrospinal fluid specimen with five or more white blood cells/high-power field and lymphoblasts seen on cytospin preparation.
B-Precursor Cell ALL
The B-precursor cell phenotype is present in 80% to 85% of children with ALL. These leukemia cells are characterized by reactivity with monoclonal antibodies specific for B-cell–associated antigens (e.g., CD9, CD19, CD22, and CD79a) and are distinguished from mature B-cell ALL by the absence of surface immunoglobulin. In the vast majority (80% to 90%) of B-precursor cell ALL, the cells express CD10, also known as the common ALL antigen (CALLA).
Over 90% of cases of B-precursor cell ALL have evidence of immunoglobulin gene rearrangements, predominantly involving the immunoglobulin heavy chain (IgH). Of note, T-cell receptor (TCR) gene rearrangements also may be observed in B-precursor cell ALL and IgH gene rearrangements have been observed in T-cell ALL. These leukemia-specific clonal IgH and TCR gene rearrangements have been used to quantify submicroscopic levels of residual leukemia in patients using polymerase chain reaction (PCR)-based assays.
The presence of intracytoplasmic immunoglobulin (cIg) and various cell surface markers has been used to distinguish different subsets of B-precursor cell ALL based on their level of differentiation. These subsets include pro-B ALL (3% to 4% of pediatric patients), early pre-B ALL (60% to 70% of pediatric patients), and pre-B ALL (20% to 30% of pediatric patients):
- •
Pro–B-cell ALL, believed to be derived from a very immature B-cell precursor, is characterized by CD10-negative immunophenotype and the absence of cIg. It is most frequently observed in infants with ALL, especially those with abnormalities involving chromosome 11q23 ( MLL [also known as KMT2A ] gene locus).
- •
Early pre–B-cell ALL is the most common subtype of B-precursor cell ALL in children. It is believed to be derived from a more mature B-cell precursor than pro-B ALL. Early pre–B cells frequently express CD10 but still lack cIg. CD10 (CALLA)-positive leukemias without cytoplasmic immunoglobulin are also referred to as common ALL (c-ALL).
- •
Pre–B-cell ALL is characterized by the presence of cIg. It is believed to derive from an intermediate B-precursor cell, more mature than those lacking cIg (early pre–B cells) but not as mature as those with surface immunoglobulin (mature B cells). Like early pre–B cells, pre–B lymphoblasts typically express CD10 and human leukocyte antigen (HLA)-DR. Approximately 25% of cases of pre–B-cell (cIg+) ALL have the chromosomal translocation t(1;19)(q23;p13), which fuses the TFPT (formerly E2A ) gene on chromosome 19 with the PBX1 gene on chromosome 1. This translocation is observed in only 1% of cases of early pre-B (or common) ALL.
Mature B-Cell ALL
Mature B-cell ALL accounts for 1% to 2% of childhood ALL cases. It is characterized by the presence of surface immunoglobulin, most often IgM, which is monoclonal for κ or λ light chains. The cells generally express other B-cell antigens, including CD19, CD20, and HLA-DR. Morphologically, the blasts tend to be classified as FAB L3, with intensely staining cytoplasmic basophilia similar to that of erythroblasts. Cases of mature B-cell ALL without L3 morphologic features have been reported but are rare. Almost all cases of mature B-cell ALL are associated with one of three nonrandom chromosomal translocations—t(8;14)(q24;q32) or, less commonly, t(2;8)(p12;q24) or t(8;22)(q24;q11). The chromosomal breakpoints involve the MYC oncogene on chromosome 8 and the genes for the IgH (chromosome 14), κ light chain (chromosome 2), or λ light chain (chromosome 22). Mature B-cell ALL is clinically indistinguishable from disseminated Burkitt lymphoma and is much more successfully treated with regimens used for that disease than when treated with conventional childhood ALL therapy.
T-Cell ALL
The T-cell immunophenotype is present in 10% to 15% of children with ALL. Compared with B-precursor cell ALL, T-cell ALL is more frequently associated with older age at diagnosis, higher presenting leukocyte counts, and bulky extramedullary disease (including lymphadenopathy, hepatosplenomegaly, overt central nervous system [CNS] leukemia, and an anterior mediastinal thymic mass). Historically, patients with T-cell ALL had an inferior outcome compared with those with B-precursor cell ALL, although this prognostic difference is not observed when more intensive regimens are used. However, the time period in which relapses occur differs between the two immunophenotypic subtypes, with earlier relapses observed more frequently in T-cell ALL.
T-cell ALL can be subclassified using monoclonal antibodies that recognize surface antigens expressed during discrete stages of normal T-cell development. Most of the phenotypically defined subsets of T-cell ALL have limited clinical relevance. However, gene expression profile studies have identified a biologically distinctive subset of T-cell ALL that, in retrospective analysis, was associated with a high risk for treatment failure. These patients were noted to have a distinctive “early T-cell precursor” phenotype consisting of weak expression of CD5 and coexpression of stem cell or myeloid markers. Similarly, another retrospective study using array comparative genomic hybridization (CGH) and quantitative DNA-PCR techniques identified the absence of V(D)J recombination at the TCRγ locus, a feature that is characteristic of early T-cell precursor cells, as a predictor of early treatment failure in patients with T-cell ALL. Thus, T-cell ALL patients with features of development arrest at the earliest stages of T-cell development appear to have an inferior outcome.
Myeloid Antigen Coexpression
In some instances, individual leukemic cells simultaneously express lymphoid and myeloid surface antigens. Up to 20% of cases of ALL (i.e., with lymphoid surface antigen predominance) also coexpress myeloid antigens. Myeloid antigen coexpression is associated with young age at diagnosis (<12 months), pro–B-cell (CD10−) immunophenotype, and certain chromosomal abnormalities, such as rearrangements of chromosome 11q23 (frequently observed in infants with ALL), the Philadelphia (Ph) chromosome [t(9;22)], and the ETV6-RUNX1 [t(12;21)] fusion. Older studies suggested that patients with biphenotypic ALL had a worse prognosis, but more recent reports have indicated that myeloid antigen coexpression lacks independent prognostic significance. Cases of ALL with myeloid antigen coexpression should be distinguished from leukemia of ambiguous lineage, an uncommon entity in which the predominant lineage (lymphoid or myeloid) cannot be determined by immunophenotype and histochemistry. The World Health Organization classification system requires that cells express myeloperoxidase or evidence of monocytic differentiation, along with either T-cell antigen CD3 (cytoplasmic or surface) or B-cell surface antigens (CD19, CD79a, CD22, and CD10).
Molecular Pathogenesis
Lymphocytes arise from hematopoietic stem cells through a series of proliferation and differentiation steps that result in the formation of mature B or T lymphocytes, each expressing a specific immunoglobulin or TCR capable of responding to a narrow range of foreign antigens. The development of a fully competent adaptive immune system is dependent on the generation of a population of mature B and T lymphocytes with extraordinary diversity in antigen specificity, which collectively provide the ability to mount an effective immune response against a broad range of pathogens. This diversity in antigen receptor specificity is achieved through an elaborate process of somatic mutagenesis, including V(D)J recombination and somatic hypermutation, designed to introduce unique variations in the immunoglobulin or TCR gene structure of each individual developing lymphocyte. Despite their carefully regulated nature, these programmed mechanisms of somatic mutagenesis can go awry and introduce somatic mutations at other genomic loci. Mutations in oncogenes or tumor suppressors acquired during lymphocyte development can result in the aberrant proliferation and differentiation arrest that are characteristic of ALL.
Although a comprehensive review of the molecular pathogenesis of ALL is beyond the scope of this chapter, we highlight here select molecular lesions that have proven particularly informative to our understanding of the pathobiology of ALL and review in more detail the molecular pathogenesis of three particularly high-risk subtypes of ALL: BCR-ABL, hypodiploid, and early T-cell progenitor ALL.
Transcriptional Regulators
ETV6-RUNX1 Fusion Genes in B-Precursor Cell ALL
In 20% to 25% of childhood ALL cases a cryptic t(12;21) translocation results in expression of a fusion ETV6-RUNX1 (also known as TEL-AML1 ) translocation. ETV6 and RUNX1 are transcription factors that are each required for normal hematopoiesis, and both are involved in other leukemia-related translocations. The t(12;21) translocation results in the fusion of the HLH domain of ETV6 to almost all of RUNX1 . The mechanisms through which ETV6-RUNX1 fusions drive leukemogenesis remain incompletely understood, but recent work has shown that expression of this fusion oncoprotein in hematopoietic stem cells markedly increases their numbers and promotes their transformation after chemical mutagenesis. Interestingly, ETV6-RUNX1 translocations can often be detected in prenatal blood specimens collected from children who went on to develop ETV6-RUNX1 –rearranged ALL many years later, indicating that it is likely the initiating mutation in ALL. However, this translocation is not sufficient for leukemogenesis because the incidence of detectable ETV6-RUNX1 fusions in normal neonatal blood is 100-fold greater than the incidence of leukemia with this translocation. Several studies have shown that the presence of an ETV6-RUNX1 translocation is associated with a favorable prognosis in childhood ALL. Indeed, ETV6-RUNX1 expression is associated with decreased expression of the ABCB1 (also known as MDR1 ) multidrug resistance gene and of genes involved in purine metabolism, which may explain the particular sensitivity of these cases to mercaptopurine and methotrexate.
MLL Fusion Genes in B-Precursor Cell ALL
Chromosomal translocations resulting in the fusion of the amino-terminus (N-terminus) of the MLL gene on 11q23 to the carboxy-terminus (C-terminus) of one of more than 60 different translocations partners occur in 70% of infant ALL, in 10% of AML, and in the majority of secondary AML cases that are induced by prior treatment with topoisomerase II inhibitors. The presence of MLL translocations is associated with inferior outcomes despite aggressive chemotherapy in ALL, as discussed later in this chapter. MLL is the human ortholog of Drosophila trithorax , which positively regulates expression of multiple genes of the HOXA cluster and acts antagonistically to the polycomb group of proteins. The breakpoints of MLL translocations cluster between exons 5 and 11 of MLL , resulting in expression of fusion oncoproteins that retain the N-terminal domains of the protein that bind DNA in a sequence-nonspecific manner, whereas the C-terminal domains of MLL that mediate association of the wild-type protein with its endogenous chromatin modification complex and its Histone H3 lysine-4–specific N -methyltransferase activity are invariably lost. Instead, the C-terminus of MLL fusion oncoproteins is provided by one of more than 60 different translocation partners, with common translocations such as the t(4;11), t(9;11), and t(11;19)(q23;p13.3) resulting in the in-frame fusion of MLL to the C-terminal domains of AF4 (also known as AFF1 ), AF9 (also known as MLLT3 ), and ENL (also known as MLLT1 ), respectively. Expression of MLL fusion oncoproteins in murine hematopoietic precursors induces leukemia, an effect that is dependent on MLL fusion oncoprotein–driven overexpression of several genes of the HOXA cluster.
Until recently, the mechanism through which the C-terminal fusion partners of MLL contributed to leukemogenesis was unclear, because there were no apparent sequence similarities between the fusion partners involved. However, recent work has shown that several MLL translocation partners are functionally linked through their association in protein complexes that regulate transcriptional elongation. MLL fusion oncoproteins bind at least three distinct complexes, including the polymerase-associated factor complex, the positive transcription elongator factor (pTEFb) complex, and the DOT1L histone methyltransferase complex. The DOT1L methyltransferase is required for the maintenance of MLL-driven leukemias but appears to be less important for normal hematopoiesis, thus suggesting a therapeutic window for targeting DOT1L in MLL-rearranged ALL. Indeed, a small molecule DOT1L inhibitor has recently been developed and demonstrates promising preclinical activity in murine models of MLL-rearranged leukemia, raising the possibility that such a therapeutic strategy could improve the prognosis for patients with MLL-rearranged ALL.
MYC Translocations in Mature B-Cell ALL
Chromosome translocations that place the coding region of the MYC protooncogene under the control of gene regulatory elements of one of the immunoglobulin receptor genes, such as the t(8;14), t(2;8) or t(8;22), lead to aberrant overexpression of the MYC oncogenic transcription factor. MYC overexpression is potently oncogenic in a wide range of experimental systems. Although MYC has long been believed to be a traditional transcription factor that regulates a specific set of target genes, recent work has revealed that MYC accumulates at the promoters of essentially all transcriptionally active genes and acts as a nonlinear amplifier of existing gene expression programs. Thus, rather than inducing expression of a specific gene expression program, these findings suggest that MYC instead acts to lock a cell’s gene expression program into its existing form, which presumably can be oncogenic when it occurs in a cell that is in a highly proliferative state that normally should be transient. MYC translocations are characteristically found in mature B-cell leukemia, which is best considered a disseminated form of Burkitt lymphoma that responds poorly to standard ALL therapy. Outcomes for these patients are much improved with treatment regimens specifically designed for high-grade B-cell non-Hodgkin lymphomas.
Activating NOTCH1 Mutations in T-Cell ALL
NOTCH1 encodes an unusual cell surface receptor that, on activation by ligand binding to its extracellular domain, undergoes two distinct proteolytic cleavage events, culminating in release of the intracellular domain of NOTCH1 (ICN) into the cytoplasm. The ICN protein subsequently translocates to the nucleus, where it functions as a transcription factor that generally activates expression of its targets. NOTCH1 is required at multiple stages of T-cell development, and its ectopic activation in murine T-cell progenitors is highly leukemogenic. Although NOTCH1 can be activated by chromosomal translocations in very rare cases of T-cell ALL, more than 50% of T-cell ALL cases harbor activating NOTCH1 mutations, and these occur in all of the molecular subtypes of T-cell ALL. Activating NOTCH1 mutations typically occur either as missense mutations in the heterodimerization domain, where they promote ligand-independent proteolytic activation of the ICN domain, or as truncating or frameshift mutations in the C-terminal PEST domain of ICN, which lead to ICN protein accumulation as a result of impaired proteasomal degradation. The pathogenic role of NOTCH1 in T-cell ALL is mediated largely by its role as a transcriptional activator of two key downstream targets, MYC and HES1 .
One of the key proteolytic cleavage events required for NOTCH1 activation is mediated by the γ-secretase complex. Treatment of NOTCH1-mutant T-cell ALL cell lines with γ-secretase inhibitors leads to modest reductions in cell proliferation in a subset of cell lines, but a number of NOTCH1-mutant T-cell ALL cell lines fail to respond to these inhibitors. The mechanisms of resistance to NOTCH1 inhibition have been an area of intense investigation, and recent work has revealed that inactivation of the FBXW7 tumor suppressor, an E3 ubiquitin ligase that targets both ICN and MYC for proteasomal degradation, is one mechanism of resistance to γ-secretase inhibitors. Additionally, resistance to inhibition of NOTCH1 or its downstream target MYC can be mediated by PTEN inactivation and resultant AKT activation in T-cell ALL, thus suggesting the potential therapeutic utility of AKT pathway inhibitors as a strategy to reverse resistance to γ-secretase inhibitor. Moreover, glucocorticoid resistance in T-cell ALL has been shown to be reversible by γ-secretase inhibitors, and it appears that glucocorticoid therapy can ameliorate the intestinal toxicity of γ-secretase inhibitors. Taken together, these findings provide a compelling rationale for clinical trials combining glucocorticoids with small molecule inhibitors of NOTCH1 and AKT.
Growth Factor Signaling
Cytokine Receptor Signaling
Cytokine receptor signaling plays key roles in normal lymphocyte development, with inactivating mutations in key cytokine receptor subunits (e.g., interleukin-2 receptor γ [ IL2RG ] and interleukin-7 receptor [ IL7R ]) or their downstream effector kinases (e.g., Janus kinase-3 [ JAK3 ]), resulting in severe lymphopenia and the lack of a functional adaptive immune system characteristic of severe combined immunodeficiency. Conversely, aberrant activation of cytokine receptor signaling has recently been implicated in the molecular pathogenesis of ALL.
Cytokine receptor–like factor 2 (CRLF2) encodes a cytokine receptor subunit that heterodimerizes with IL7R to form the receptor for the cytokine TSLP (thymic stromal lymphopoietin), which is a proinflammatory cytokine secreted by endothelial cells that drives lymphocyte proliferation. CRLF2 rearrangements leading to aberrant overexpression have recently been described in B-precursor cell ALL, typically as a result of an intrachromosomal deletion leading to a P2RY8-CRLF2 fusion, or translocations of the IgH locus to CRLF2 . CRLF2 rearrangements are found in 5% to 10% of B-precursor cell ALL cases but in more than 50% of patients with Down syndrome–associated ALL, strongly suggesting that these collaborate with trisomy 21 in B-lymphoblast transformation. CRLF2 rearrangements are often associated with activating mutations of Janus kinase 2 (JAK2) , and CRLF2 overexpression and mutated JAK2 cooperate to induce cytokine-independent growth to Ba/F3 pro-B cells.
IL7R encodes a cytokine receptor subunit that can heterodimerize with the IL2Rγ (the common γc chain) to form IL7R, or with CRLF2 to form the TSLP receptor. Recent work has identified activating IL7Rα point mutations in 9% of cases of T-cell ALL, as well as in the “BCR-ABL–like” subset of B-precursor cell ALL (discussed in more detail later). Interestingly, most of these mutations result in the introduction of a novel cysteine residue into the extracellular domain of IL7Rα, and aberrant signaling results because disulfide bond formation between these mutant cysteines can allow mutant IL7Rα proteins to aberrantly dimerize, leading to activation of downstream signaling pathways independent of ligand or of the common γc chain. The potential therapeutic utility of targeting aberrant oncogenic signaling downstream of mutationally activated cytokine receptors is an area of active investigation.
PTEN and the PI3K-AKT Axis
The PI3K -AKT oncogenic pathway, which is negatively regulated by the PTEN tumor suppressor, mediates prosurvival and proliferative signaling downstream of activated growth factor receptors and is aberrantly activated in a broad range of human cancer subtypes. Recent work has revealed that nearly half of T-cell ALL cases harbor mutational activation of oncogenic signaling through this pathway, most often as a result of truncating mutations or deletions of the PTEN tumor suppressor, but activating mutations of PI3K and AKT genes also occur. PTEN deletions are a strong predictor of early treatment failure in T-ALL cases and PTEN inactivation has been shown to induce resistance to NOTCH1 or MYC inhibition in zebrafish and murine models of T-cell ALL. These findings highlight the potential application of small molecule inhibitors of the PI3K-AKT pathway for high-risk T-cell ALL.
Molecular Pathogenesis of Select High-Risk Subtypes
BCR-ABL and BCR-ABL –like ALL
The discovery of the Ph chromosome, which arises from the t(9;22)(q34;q11) translocation resulting in expression of a BCR-ABL fusion oncoprotein, was a major milestone in molecular oncogenesis that provided compelling support for the hypothesis, first put forth by Boveri in 1914, that cancer arises due to a genetic lesion in a single cell, resulting in outgrowth of a clonal population. BCR-ABL translocations are pathognomonic of chronic myeloid leukemia and are also found in 4% to 5% of childhood and 25% of adult cases of B-precursor cell ALL. The translocation underlying formation of the Ph chromosome results in expression of a BCR-ABL fusion oncoprotein with constitutively active tyrosine kinase activity, which transforms cells at least in part due to activation of the RAS-MAPK pathway, PI3K, and JUN-kinase pathways. Although historically associated with an extremely poor prognosis, hematopoietic stem cell transplantation (HSCT) proved to be the first highly effective therapeutic modality for this high-risk subtype of ALL. Additionally, the development of imatinib mesylate, a small molecule inhibitor of oncogenic signaling by the BCR-ABL kinase, has opened novel therapeutic opportunities for the management of Ph chromosome–positive ALL. Although single-agent therapy with imatinib leads to rapid development of resistance in Ph chromosome–positive ALL, pilot trials of the combination of conventional chemotherapy with imatinib or other tyrosine kinase inhibitors have been promising.
Although the poor prognosis of BCR-ABL ALL was traditionally believed to be due to expression of the BCR-ABL oncoprotein, recent work has shown that this subtype of ALL is also characterized by inactivation of the DNA-binding protein Ikaros, which is encoded by the IKZF1 gene. IKZF1 deletions have been identified in up to 15% of BCR-ABL–negative pediatric ALL cases and have been shown to be an independent predictor of adverse outcome. These cases have been classified as “BCR-ABL–like” ALL on the basis of similarities in gene expression profiles with Ph chromosome–positive ALL. Recent work has revealed that most of these “BCR-ABL–like” cases harboring Ikaros alterations, but lacking BCR-ABL translocations, commonly harbor mutational activation of tyrosine kinase signaling as a result of translocations or activating point mutations of receptor or cytoplasmic kinases, such as PDGFRB, CRLF2, EPOR, IL7R, FLT3, JAK2, and ABL1. Importantly, preclinical studies have demonstrated the potential therapeutic utility of targeting the aberrantly activated kinase involved for at least some of these cases.
Hypodiploid ALL
The presence of hypodiploidy (<44 to 45 chromosomes) portends a poor prognosis in B-precursor cell ALL, with event-free survival (EFS) rates less than 40%. Alterations in whole-chromosome number (aneuploidy) are very common in human cancer and can be oncogenic. However, the mechanisms through which hypodiploidy contributes to leukemogenesis in B-precursor cell ALL remain largely unknown.
A recent genomic study has shed key insights into the molecular pathogenesis of hypodiploid ALL. Both copies of chromosome 21 were always retained in each of the 124 cases of hypodiploid ALL cases analyzed, which, given that trisomy 21 is one of the most common genetic alterations in B-precursor cell ALL, highlights the pathogenic role of additional copies of genes on chromosome 21 in B-precursor cell ALL.
Rather than representing a single subtype of ALL, hypodiploid ALL instead harbors distinct biologic subsets, including low hypodiploidy (32-39 chromosomes) and near haploidy (24-31 chromosomes), which differ strikingly on a molecular basis.
- •
Near-haploid ALL (24 to 31 chromosomes): Seventy percent of these cases are characterized by genetic lesions leading to activation of growth factor signaling pathways, including NRAS, KRAS, NF1, MAPK, FLT3, and PTPN11. Notably, deletions of tumor suppressors such as NF1 and PTPN11 were typically homozygous as a result of hypodiploidy, and loss of heterozygosity of tumor suppressors provides one mechanism through which hypodiploidy can contribute to oncogenesis. These data support the potential therapeutic utility of targeting these kinases, or their key downstream effectors, although such interventions await further preclinical investigation.
- •
Low-hypodiploid ALL (32 to 39 chromosomes): In marked contrast to near-haploid ALL, almost all cases of low-hypodiploid ALL harbored mutations of the TP53 tumor suppressor, whereas mutations of growth factor signaling pathways were rare in this subset. Mutations of TP53 were present in remission bone marrow specimens in almost half of pediatric cases, strongly suggesting a germline origin consistent with the Li-Fraumeni syndrome; indeed, germline transmission was confirmed in one kindred. Given that the induction of aneuploidy triggers TP53- dependent cell cycle arrest, these data support a model in which TP53 inactivation occurs early in the pathogenesis of low-hypodiploid ALL, thus allowing the subsequent acquisition of hypodiploidy to be tolerated by these cells. However, it remains entirely unclear why TP53 mutations should be so rare in other ALL subtypes with striking degrees of aneuploidy, including near-haploid and hyperdiploid ALL. Low-hypodiploid cases were also associated with a high frequency of inactivating mutations of the RB1 tumor suppressor, as well as a very high frequency of mutations involving Ikaros family members, particularly IKZF2 (encoding Helios), which was mutated in more than 50% of cases.
A subset of cases with an apparently hyperdiploid chromosome complement was found to in fact represent “masked” hypodiploidy in which the hypodiploid genome has undergone reduplication, based on loss of heterozygosity apparent on the single-nucleotide polymorphism arrays performed. Although these cases might be classified clinically as hyperdiploid ALL (which has a favorable prognosis), masked hypodiploid ALL cases are indistinguishable from the respective hypodiploid ALL subtype (low-hypodiploid or near-haploid) based on gene expression profiling and pattern of oncogenic lesions, thus suggesting the need for clinical tests to distinguish this subtype from cases of “typical” hyperdiploidy, which has a favorable prognosis. Given that chromosomes 4 and 17 are lost in almost all cases of hypodiploid ALL, the presence of these trisomies may help to distinguish cases of true hyperdiploidy from masked hypodiploidy.
Early T-Cell Progenitor T-Cell ALL
Recent work has revealed that differentiation arrest at the earliest stages of T-cell development, defined either by absence of biallelic TCRγ deletion (ABD; indicating failure to complete V(D)J recombination) or by an early T-cell progenitor gene expression profile pattern, identifies an overlapping subset of T-cell ALL cases at very high risk for treatment failure. These cases are associated with aberrant oncogenic signaling through the RAS-MAPK, PI3K-AKT, and JAK-STAT pathways, typically owing to associated mutations in RAS , FLT3 , IL7R , BRAF , and JAK kinases, as well as deletions of the PTEN tumor suppressor. These cases are defined as T-cell ALL based on expression of cytoplasmic CD3 and harbor mutations in defining T-cell ALL oncogenes such as NOTCH1 . However, these cases also harbor additional mutations that are typically seen in AML, myelodysplastic, and myeloproliferative syndromes, such as RUNX1 , ETV6 , EED , EZH2 , SUZ12 , DNMT3A , IDH1 , and IDH2 . Moreover, gene expression analyses have shown that early T-precursor T-cell ALL harbors similarities to normal hematopoietic stem cells and AML leukemic stem cells. The identification of effective targeted therapies is a subject of active investigation.
Clinical Manifestations
ALL may present insidiously or acutely, as an incidental finding on a routine blood cell count of an asymptomatic child, or as a life-threatening hemorrhage, infection, or episode of respiratory distress. Common presenting symptoms include fever, pallor, bruising, petechiae, bone pain (presumably secondary to stretching of the periosteum or joint capsule by leukemic infiltration), and limp.
Although ALL is primarily a disease of the bone marrow and peripheral blood, any organ or tissue may be infiltrated by the abnormal cells. Such infiltration may be clinically apparent by physical examination. At initial diagnosis, 30% to 50% of children have enlargement of the liver or spleen, with organs palpable more than 4 cm below the costal margins (see Table 50-1 ). Lymphadenopathy caused by leukemic infiltration is an equally frequent presenting sign. Generally, the degree of organ infiltration correlates with peripheral blood blast count, thus reflecting the total leukemic mass. Leukemic invasion of tissues may, however, be occult and detectable only by histologic sampling.
Peripheral Blood
Clinical laboratory data often provide a broad spectrum of abnormal findings at the time of diagnosis of leukemia. Anemia, abnormal leukocyte counts and differential, and thrombocytopenia are common (see Table 50-1 ). However, some children with ALL may have normal peripheral blood cell counts at the time of diagnosis, even when the bone marrow is replaced by leukemic cells.
The red blood cells are usually normochromic and normocytic. Failure of erythroid production is manifested by a low reticulocyte count. Peripheral blood smear may reveal teardrop forms and nucleated red cells, consistent with marrow invasion.
Platelet counts may vary from normal to extremely low. Most children have fewer than 100,000 cells/mm 3 at presentation, and the platelets are usually of normal size. In contrast to idiopathic thrombocytopenic purpura (ITP), thrombocytopenia at the time of ALL diagnosis is usually accompanied by other hematologic or physical manifestations of leukemia.
There is a wide range of leukocyte counts observed at the time of diagnosis, from extremely low to more than 1 million cells/mm 3 . Approximately 20% of children with ALL present with leukocyte counts of more than 50,000 cells/mm 3 . Even with high presenting leukocyte counts, absolute neutropenia is common. Blast forms may or may not be present on routine smears of peripheral blood. Even when present, peripheral blast cells may be misleading, and the definitive diagnosis of leukemia cannot be made from the morphologic examination of the peripheral blood. For example, myeloblasts may be detected in the peripheral blood when the marrow has been infiltrated by various disorders, including osteopetrosis, myelofibrosis, granulomatous infections, sarcoidosis, metastatic tumor, and even ALL. Additionally, in patients with leukemia, the morphologic appearance of leukemic blast cells in the peripheral blood may differ from that of the marrow. Peripheral blood flow cytometry may be useful in distinguishing malignant blasts from “normal” myeloblasts, and differentiating ALL from AML, in patients with high presenting leukocyte counts.
Hypereosinophilia has been described in association with ALL. The eosinophilia may precede the diagnosis of ALL, appear at presentation, and then disappear with successful remission induction. Eosinophilia in B-precursor cell ALL has been associated with the t(5;14)(q31;q32) translocation, involving the IL3 gene on chromosome 5 and the IgH gene (IGH) on chromosome 14.
In regard to hyperleukocytosis, approximately 10% of children with ALL present with extremely high leukocyte counts (>100,000 cells/mm 3 ) ( Table 50-2 ). In patients with markedly elevated leukocyte counts, blood flow in the microcirculation can be impeded by intravascular clumping of the poorly deformable blasts. This may result in local hypoxemia, endothelial damage, hemorrhage, and infarction, especially in the CNS and lung. Clinically significant leukostasis, resulting in intracerebral hemorrhage or respiratory failure, is more common with hyperleukocytosis in AML than in ALL. In one series, neurologic or pulmonary symptoms or both were observed in 5% to 10% of children with ALL and a presenting leukocyte count higher than 200,000 cells/mm 3 and CNS hemorrhage was diagnosed in 2% of those patients (all of whom had leukocyte counts higher than 400 × 10 9 /L). Treatment of hyperleukocytosis consists of vigorous intravenous hydration. Red blood cell transfusions should be administered cautiously to avoid increasing whole blood viscosity and worsening symptoms. For patients with very high presenting leukocyte counts and symptomatic leukostasis, leukapheresis or exchange transfusion may prevent hyperviscosity and leukemia cell lysis–related problems.
| Presenting Feature | Age (yr) | ||
|---|---|---|---|
| < 1 (2.5% of Patients) | 1 to < 10 (79% of Patients) | 10-18 (18.5% of Patients) | |
| Gender | |||
| Male | 52% | 56% | 61% |
| Female | 48% | 44% | 39% |
| WBC count ≥100 × 10 9 /L | 59% | 10% | 15% |
| Phenotype | |||
| B-precursor cell | 96% | 90% | 73% |
| T-cell | 4% | 10% | 27% |
| CNS-positive * | 21% | 2.3% | 3.9% |
| High hyperdiploidy † | 1.5% | 27% | 15% |
| ETV6-RUNX1 fusion † | 4.5% | 27% | 11% |
| t(9;22) BCR-ABL † | 0% | 2.2% | 5.1% |
* CNS-3, diagnostic cerebrospinal fluid specimen with five or more WBCs/high-power field and lymphoblasts seen on cytopsin preparation.
Disseminated intravascular coagulation is infrequently observed in patients with ALL at diagnosis. It is more common in T-cell ALL as a result of thromboplastic substances released from T lymphoblasts. It also has been reported in patients with the uncommon t(17;19) translocation, associated with the B-precursor cell immunophenotype. Disseminated intravascular coagulation should be treated promptly with infusions of fresh-frozen plasma and cryoprecipitate.
Bone Marrow
Evaluation of the bone marrow by aspiration and biopsy is essential for making the diagnosis of leukemia. The marrow specimen is usually hypercellular and characterized by a homogeneous population of cells. In most cases of ALL, the marrow generally has more than 50% blasts.
A bone marrow aspirate may be difficult to obtain at the time of diagnosis. This is usually caused by the density of blast forms in the marrow, but it may be caused by marrow fibrosis or necrosis. In such cases, a diagnosis can be made by bone marrow biopsy. “Touch preps” of the biopsy specimen can be helpful in elucidating morphology when aspiration is not successful.
Differences may occur in leukemic involvement found in marrow aspirates derived from widely separated sites. Patchy marrow involvement of leukemic infiltration has been reported at diagnosis and relapse. If clinical findings suggest leukemia, but a single-site bone marrow specimen is nondiagnostic, the marrow should be sampled at additional sites.
The distinction between ALL with lymph node involvement and non-Hodgkin lymphoblastic lymphoma with bone marrow invasion (stage IV) is arbitrary. Commonly the disease is classified as ALL when there are more than 25% lymphoblasts in the marrow and as stage IV non-Hodgkin lymphoblastic lymphoma when there are fewer than 25% lymphoblasts in the marrow. Although the lymphoblasts of ALL and lymphoblastic lymphoma appear morphologically and immunophenotypically indistinguishable, gene expression profiling studies have suggested that there may be differences in underlying biology between these two diagnoses. However, the distinction may not be clinically important because advanced-stage lymphoblastic lymphoma and ALL respond similarly to intensive ALL-type therapy.
Extramedullary Leukemia
Although marrow replacement is the major cause of symptoms of leukemia, extramedulllary involvement may also contribute to presenting signs and symptoms.
CNS Manifestations
Up to 20% of children will have blast cells visible on a cytocentrifuged cerebrospinal fluid (CSF) specimen at diagnosis. CSF lymphoblasts can usually be identified by the use of cytocentrifugation and Wright-Giemsa staining. Such morphologic evaluation is necessary to distinguish the pleocytosis of leukemic meningitis from that induced as a result of intrathecal chemotherapeutic agents (arachnoiditis) or from CNS infections.
The vast majority of children with detectable CSF lymphoblasts at initial diagnosis do not have any symptoms related to CNS involvement. Symptomatic patients may present with diffuse or focal neurologic signs and symptoms, including manifestations of increased intracranial pressure (vomiting, headache, papilledema, and lethargy), seizures, and nuchal rigidity. Cranial nerve palsies can occur, with the facial nerve being the most frequently involved.
Intracranial hemorrhage in patients with high presenting leukocyte counts is uncommon in ALL but can lead to severe and devastating neurologic consequences. Clinically significant spinal cord involvement has been reported in ALL, manifesting as a localized epidural leukemic infiltrate compressing the cord.
Anterior Mediastinal Mass
Leukemic infiltration of the thymus appears as an anterior mediastinal mass on a chest radiograph. It is observed in about 10% of newly diagnosed patients and is almost always associated with T-cell immunophenotype (see Table 50-1 ). Leukemic infiltration of the mediastinal structures may cause life-threatening tracheobronchial or cardiovascular compression. Pleural effusion may also be associated with thymic enlargement, which can exacerbate respiratory distress. Prompt initiation of systemic chemotherapy (e.g., corticosteroids) is necessary to handle such emergencies and, rarely, emergent radiation may be indicated.
Genitourinary Tract Manifestations
Testicular Enlargement.
The clinical presentation of testicular ALL is a painless enlargement of one or both testes. Clinically detectable testicular leukemia is uncommon at diagnosis, occurring in 1% to 2% of boys, and does not appear to have prognostic significance. Occult testicular involvement at diagnosis is more common, especially in the presence of a high tumor burden. In a study in which testicular biopsies were performed in boys with newly diagnosed ALL, approximately 20% had microscopic leukemic involvement. However, this finding does not appear to have any prognostic significance, and routine testicular biopsies at the time of diagnosis to document occult disease are not recommended.
Other Genitourinary Tract Sites of Involvement.
Ultrasonographically enlarged kidneys have been observed in children with ALL at the time of diagnosis and are believed to be primarily caused by leukemic infiltration. Renal enlargement in acute leukemia also may be related to hyperuricemia, hemorrhage, and pyelonephritis. Hypertension is more commonly associated with the treatment of leukemia, especially with the prolonged use of corticosteroids, than with renal involvement. Occasionally, urolithiasis is observed at presentation or during therapy for ALL. Most renal stones in ALL patients are calcium-based in composition and occur most frequently during phases of therapy that include corticosteroids.
Priapism is rare and usually associated with an elevated white blood cell count. The pathogenesis may be caused by involvement of sacral nerve roots, or it may be related to mechanical obstruction of the corpora cavernosa and dorsal veins by leukemic infiltration or leukostasis.
Bone and Joint Manifestations
Bone pain, joint pain, and limp are common presenting symptoms in childhood ALL. Bone pain may be the result of direct leukemic infiltration of the periosteum, periosteal elevation of underlying cortical disease, bone infarction, or expansion of the marrow cavity by the leukemic cells. Pain and swelling of the joint are less frequent but may be presenting manifestations of disease and can initially cause confusion in the diagnosis. Migratory joint pain accompanied by swelling and tenderness can be misdiagnosed as juvenile rheumatoid arthritis (JRA) or rheumatic fever.
Up to 25% of children with ALL have characteristic radiographic changes, such as osteopenia and fracture at diagnosis, including vertebral compression fractures. Some patients have radiographic changes in the absence of bone pain, whereas others have bone pain unaccompanied by radiographic changes. Radiographic changes are most often seen in the long bones, especially around the areas of rapid growth (e.g., the knees, wrists, and ankles), and include subperiosteal new bone formation, transverse metaphyseal radiolucent bands, osteolytic lesions involving the medullary cavity and cortex, diffuse demineralization, and transverse metaphyseal lines of increased density (growth arrest lines). The latter probably represent regions of growth arrest during active phases of the disease rather than direct infiltration by leukemic cells.
Gastrointestinal Manifestations
Hepatosplenomegaly is a common feature at the time of diagnosis. Severe hepatic dysfunction, manifested as hyperbilirubinemia, presumably from leukemic infiltration, has been observed at diagnosis in some children with ALL and can complicate initial therapy, because many induction agents are hepatically metabolized or hepatotoxic. A short course of corticosteroid monotherapy before the initiation of multiagent induction chemotherapy may improve hyperbilirubinemia.
Ocular Manifestations
Ophthalmic manifestations of leukemia can be observed in more than a third of all newly diagnosed patients. Leukemia can involve almost all ocular structures. Retinal hemorrhages, the most frequent ocular abnormality, are presumably caused by thrombocytopenia or anemia. However, local infiltration of the capillary vessel walls with subsequent rupture and hemorrhage also may occur, especially in patients with very high leukocyte counts. Ocular motor palsies and papilledema are indicative of meningeal leukemia. Occasionally the optic nerve may be directly involved by leukemic infiltration. In such situations visual acuity may be markedly affected and patients may present with monocular blindness. Prompt radiation therapy may be necessary to salvage useful vision. Leukemic infiltration of the anterior chamber with hypopyon and iritis has also been reported and can be the first manifestation of relapse. Symptoms include conjunctival injection, photophobia, pain, blurring, and decreased vision.
Pulmonary Manifestations
Pulmonary leukostasis, which can lead to respiratory failure, can be seen at diagnosis, especially in patients with high presenting leukocyte counts. This complication is more common in AML than in ALL. Radiographic distinction among infection, leukemic infiltration, and hemorrhage may be difficult.
Dermatologic Manifestations
Skin infiltration is uncommon in childhood leukemia, with the exception of congenital leukemia (both AML and ALL). Leukemia cutis typically manifests as red or violaceous papules, nodules, or plaques.
Differential Diagnosis and Prognosis
A careful history and physical examination, together with an examination of the peripheral blood and bone marrow, result in a straightforward diagnosis of leukemia in the vast majority of cases. However, at times, ALL may present as the signs and symptoms of other conditions. These include ITP, aplastic anemia, JRA, infectious mononucleosis and other infections, and metastatic solid tumors.
Differential Diagnosis
Idiopathic Thrombocytopenic Purpura
ITP is the most common cause of the acute onset of petechiae and purpura in children. Patients with ITP usually present with isolated thrombocytopenia, often with large platelets seen on a blood smear, contrasting sharply to the more generalized blood cell abnormalities and small platelets typically observed in patients with ALL. Physical findings in ITP are usually limited to bruising or bleeding associated with thrombocytopenia. An occasional patient develops modest splenomegaly, probably caused by a recent viral infection. In most cases, the leukocyte level and differential count are unremarkable and there is no anemia unless bleeding has been substantial or an unrelated anemia is present. Bone marrow examination is rarely necessary in uncomplicated cases of pediatric ITP (i.e., children with otherwise normal blood cell counts, consistent peripheral blood smear, and absence of adenopathy, hepatosplenomegaly, or other concerning findings on physical examination). However, if the diagnosis of ITP is in question or any of these concerning findings are present, clinicians should consider marrow evaluation to rule out leukemia before starting corticosteroids to avoid partially treating occult ALL. The bone marrow aspirate is usually diagnostic because the marrow elements in ITP appear normal or show increased megakaryocytes.
Aplastic Anemia, Myelodysplasia, and Myeloproliferative Disorders
Like patients with leukemia, those with aplastic anemia, myelodysplasia, and myeloproliferative disease may present with pancytopenia and have fevers or infections associated with granulocytopenia. Lymphadenopathy and hepatosplenomegaly are unusual in aplastic anemia. Also, the radiographic skeletal changes sometimes seen in leukemia do not occur in aplastic anemia. To differentiate acute leukemia from these other disorders, marrow biopsy is mandatory and usually revealing.
Rarely, patients with ALL present with a transient pancytopenia preceding the leukemic phase. The pancytopenic period may persist for a few days or weeks and be accompanied by high fevers. Spontaneous recovery with a period of normal blood cell counts often occurs before the onset of ALL. In those cases, ALL typically becomes manifest several months after the period of aplasia. However, ALL may also develop after a period of aplasia, without any recovery of blood cell counts. Distinguishing pre-ALL pancytopenia from aplastic anemia may be difficult. Repeated bone marrow aspirates or biopsies ultimately establish the correct diagnosis by demonstrating areas of marrow that are infiltrated with lymphoblasts. The pathogenesis of the preleukemic pancytopenia prodrome is unclear; in one case report, leukemia-associated genetic lesions were identified in the aplastic marrow when it was retrospectively analyzed.
Juvenile Rheumatoid Arthritis and Connective Tissue Disease
Because ALL often presents primarily as joint or extremity complaints (especially limp, arthritis, or arthralgia), it may be confused with JRA or other autoimmune disorders. Children with JRA can manifest fever, pallor, splenomegaly, and anemia, and patients with ALL can present with a positive test for antinuclear antibody, highlighting how difficult it may be to distinguish these conditions from each other. Because of this difficulty, bone marrow aspiration should be considered before initiating corticosteroid therapy in children with presumed JRA.
Infectious Mononucleosis and Other Viral Infections
Childhood infectious mononucleosis and other viral illnesses can masquerade as leukemia. Patients may have generalized lymphadenopathy, splenomegaly, rash, fevers, and peripheral blood lymphocytosis. The atypical lymphocytes observed with acute Epstein-Barr virus (EBV) and other viral infections can sometimes be confused with peripheral leukemic blasts because they are larger than normal lymphocytes. Usually, viral infections can be differentiated from leukemia without bone marrow aspiration, but the procedure is sometimes necessary for accurate diagnosis. The detection of acute EBV infection in the blood by serology or PCR testing may be helpful in establishing the correct diagnosis.
Metastatic Solid Tumors
ALL and neuroblastoma may have similar presenting signs and symptoms, including fever, bone pain, and pancytopenia. Children with neuroblastoma frequently have malignant involvement of liver, lymph nodes, bone, or bone marrow; the marrow involvement may be extensive. In the bone marrow, neuroblasts tend to cluster and form pseudorosettes, in contrast to the diffuse involvement generally observed in ALL. Other solid tumors also rarely present as extensive marrow involvement, including rhabdomyosarcoma and Ewing sarcoma, and can initially be confused with leukemia. If questions remain after the examination of marrow morphology, other studies, such as immunophenotype and cytogenetics, may be helpful in distinguishing leukemia from a metastatic solid tumor.
Prognostic Factors
An array of clinical and biologic features have been identified as prognostically significant in childhood ALL, including age, presenting leukocyte count, immunophenotype, chromosomal abnormalities (ploidy, translocations), the presence of overt CNS leukemia at diagnosis, and the rapidity with which patients respond to initial induction chemotherapy. Ultimately the prognostic significance of any factor is treatment dependent and the importance of a particular presenting feature in predicting outcome may vary, depending on the therapy delivered to that patient.
Age
The age of patients with ALL significantly correlates with clinical outcome. In childhood ALL, infants and adolescents have a worse prognosis than patients in the intermediate age group (aged 1 to 10 years). ALL in infancy (<1 year at diagnosis) is associated with high presenting leukocyte counts, increased frequency of CNS leukemia at presentation, and a high incidence of rearrangements of the MLL gene on chromosome 11q23 (see Table 50-2 ). Even when treated with intensified regimens, infants with MLL gene rearrangements have a dismal prognosis, with long-term EFS rates ranging from 10% to 40%.
Adolescents (10 to 21 years of age) with ALL also have a less favorable outcome than children aged 1 to 10 years, although it is not as poor as that in infants. Adolescents with ALL more frequently present with “high risk” features at diagnosis, including T-cell immunophenotype, higher presenting leukocyte counts, and a lower incidence of cytogenetic abnormalities associated with a favorable prognosis, including high hyperdiploidy and the ETV6-RUNX1 gene fusion (see Table 50-2 ). Multiple retrospective studies have shown that adolescents fare better with pediatric ALL regimens than on treatments designed for adults with ALL.
Leukocyte Count
The initial peripheral blood leukocyte count is a significant predictor of treatment outcome, with worsening outcomes as the leukocyte count increases. Since 1996, based on the recommendation of the Cancer Therapy Evaluation Program of the National Cancer Institute (NCI), many investigators have considered a leukocyte count of 50,000 cells/mm 3 as the level separating patients with a higher risk for relapse from those with a more favorable prognosis.
Immunophenotype
Historically, immunophenotype was considered an important prognostic factor, with inferior outcomes observed in patients with mature B-cell and T-cell disease. However, if they are treated with more intensive regimens, children with T-cell phenotype fare as well as those with B-precursor cell disease. As noted earlier, mature B-cell ALL is more effectively treated with the same therapy used for advanced-stage Burkitt lymphoma.
Some investigators have reported prognostic differences within subsets of patients with B-precursor cell ALL. Initial studies have suggested that patients with pre–B-cell ALL (cIg+) have a worse outcome than those with early pre–B-cell ALL (cIg−). Subsequent reports indicated that the adverse outcome of patients with pre–B-cell ALL was caused by the subset with the t(1;19) translocation. However, with greater treatment intensity, even patients with this cytogenetic abnormality do not appear to have an adverse prognosis.
An early T-precursor phenotype is observed in 10% to 15% of cases of childhood T-cell ALL. This subset was initially indentified by gene expression profile analyses and is characterized by a distinctive immunophenotype (CD1a and CD8 negativity, with weak expression of CD5 and coexpression of stem cell or myeloid markers). Retrospective analyses suggested that patients with early T-precursor cell ALL have a poorer prognosis. In another retrospective study, patients with T-cell ALL with early T-precursor cell features were identified by ABD, indicating failure to complete V(D)J recombination at this locus. Like early T-precursor cell ALL patients defined by immunophenotype, T-cell ALL patients with ABD were also found to have an inferior prognosis.
In 15% to 30% of patients with newly diagnosed ALL, flow cytometry reveals coexpression of at least one myeloid antigen on the cell surface of the lymphoblasts. Myeloid antigen coexpression has been associated with several genetic abnormalities, including the ETV6-RUNX1 fusion [t(12;21)], MLL gene (11q23) rearrangements, and the Ph chromosome ( BCR-ABL rearrangement), but is almost never observed in high hyperdiploid ALL (51 to 65 chromosomes). Myeloid antigen coexpression was previously believed to be associated with an inferior outcome, but several more recent reports have indicated that it is not an independent prognostic factor.
Chromosomal Abnormalities
Several recurrent chromosomal abnormalities are important prognostic factors in childhood ALL. Two abnormalities, high hyperdiploidy (51 to 65 chromosomes or a DNA index greater than or equal to 1.16) and the cryptic t(12;21) ( ETV6-RUNX1 fusion), have been associated with a favorable prognosis. High hyperdiploidy is observed in 25% to 30% of cases of childhood ALL and is more common in younger (noninfant) children with B-precursor cell phenotype and low leukocyte counts. The most favorable outcomes in high hyperdiploid ALL patients have been associated with the presence of trisomies of chromosomes 4, 10, and 17.
The ETV6-RUNX1 fusion, which is rarely detected by karyotypic analysis, has been identified in approximately 20% of children with ALL using other more sensitive techniques, such as fluorescence in situ hybridization and the reverse-transcriptase PCR assay. Like high hyperdiploidy, the ETV6-RUNX1 fusion is more common in younger (noninfant) patients with low leukocyte counts and is observed almost exclusively in patients with B-precursor cell phenotype. Up to 80% of children with B-precursor cell ALL diagnosed between the ages of 2 and 7 years have either high hyperdiploidy or the ETV6-RUNX1 fusion, although almost never both; these two chromosomal abnormalities are almost always mutually exclusive. Several studies have indicated that children with ETV6-RUNX1– positive ALL have favorable EFS rates. When relapses are observed in children with ETV6-RUNX1– positive ALL, they tend to occur relatively late and may be more responsive to postrelapse salvage therapy.
Chromosomal abnormalities associated with an adverse prognosis include hypodiploidy (fewer than 44 or 45 chromosomes), rearrangements of the MLL gene on chromosome 11q23, and the Ph chromosome [t(9;22)]. MLL gene rearrangements are more frequent in infants than in older children with ALL. The incidence of Ph chromosome–positive ALL increases with age at diagnosis and is more common in adolescents and young adults. Patients with Ph chromosome–positive ALL appear to fare better if treated with allogeneic hematopoietic stem cell transplantation (HSCT) in first remission ; recent work has suggested that favorable outcomes may also be achieved with the combination of tyrosine kinase inhibitors and intensive chemotherapy without transplant.
Intrachromosomal amplification of the AML1 gene on chromosome 21 (iAMP21), detected in 1% to 2% of children with ALL, may also be associated with an inferior outcome. Patients with this abnormality appear to fare better with more intensified “high risk” treatment than with less intensive regimens intended for patients with “standard risk” disease. The t(1;19) translocation, observed in approximately 5% of cases of childhood ALL, had been previously associated with inferior EFS rates, but more recently conducted clinical trials have not found that this translocation is associated with an adverse prognosis. Some investigators have reported that patients with the t(1;19) translocation are at higher risk for CNS relapses, but this finding has not been confirmed by others. Chromosomal abnormalities in childhood ALL are reviewed in depth in Chapter 44 .
Overt CNS Disease at Diagnosis
Lymphoblasts in the CSF can be detected in 15% to 20% of children who present with ALL. Some children, such as those diagnosed within the first 12 months of life and those with T-cell ALL, have a higher incidence of CNS leukemia at diagnosis.
Most investigators consider the presence of overt CNS disease at diagnosis to be an adverse prognostic indicator, and these patients are usually treated with more aggressive therapy. CNS status at presentation is usually classified as CNS-1 (no blast cells), CNS-2 (fewer than five leukocytes/µL with blast cells), and CNS-3 (five or more leukocytes/µL with blast cells or cranial nerve palsy). Historically, CNS leukemia was defined as CNS-3 status (observed in approximately 5% of patients at diagnosis). CNS-3 status at diagnosis is associated with a lower probability of EFS. Several investigators have reported that patients with CNS-2 status also have a higher risk for relapse, although it does not appear to be as high as in patients with CNS-3 status at diagnosis. Intensification of CNS-directed therapy (e.g., increasing the frequency of intrathecal chemotherapeutic treatments) may abrogate the adverse prognostic significance of CNS-2 status. Thus patients with CNS-2 status at diagnosis usually receive extra doses of intrathecal chemotherapy without other changes in treatment whereas those with CNS-3 status are typically classified as higher risk and receive more intensive systemic and CNS-directed therapies.
Traumatic lumbar punctures with lymphoblasts on cytospin have also been associated with an adverse prognosis. Like patients with CNS-2 status, those with traumatic lumbar punctures with lymphoblasts at diagnosis may also benefit from additional doses of intrathecal chemotherapy.
Early Response to Induction Chemotherapy
The rapidity with which a patient responds to initial chemotherapy is a significant predictor of long-term outcome. Early response to therapy has been evaluated using morphologic measures (residual microscopic leukemia) and more sensitive techniques, such as PCR assay and flow cytometry.
Morphologic Response to Therapy.
Patients who require two or more cycles of induction chemotherapy to achieve complete remission have a much worse prognosis than those who achieve complete remission within 1 month of diagnosis. The Berlin-Frankfurt-Munster (BFM) group treats patients with 1 week of corticosteroid monotherapy (and one dose of intrathecal methotrexate) before beginning multiagent induction chemotherapy and has reported that poor peripheral blood response at the end of that week (defined as an absolute blast count of 1000/mm 3 ) is an independent predictor of adverse outcome. Others have reported that the rate of clearance of blasts from the peripheral blood after multiagent chemotherapy is also an independent predictor of relapse. Similarly, the persistence of leukemia in bone marrow specimens obtained 7 or 14 days after beginning multiagent chemotherapy strongly correlates with poor outcome, although intensification of therapy can abrogate the adverse prognostic significance of slow early response.
Minimal Residual Disease.
Minimal residual disease (MRD) evaluation involves the measurement of very low levels of leukemia using sensitive techniques, such as PCR assay or specialized multiparameter flow cytometry. Leukemic cells are identified using targets identified at diagnosis, including leukemia-specific immunophenotypes (for flow cytometry–based assays), chromosomal translocations, or lymphoblast-specific immunoglobulin or T-cell antigen receptor gene rearrangements (for PCR-based assays). Using these techniques, leukemia cells have been identified at levels as low as 1 in 1,000 to 1 in 100,000 cells.
Many studies have demonstrated that MRD levels early in therapy are a significant and independent predictor of long-term outcome. For patients achieving a morphologic remission at the end of induction therapy, those with higher levels of marrow MRD at that time point have a higher risk for relapse than those with lower or undetectable MRD. The risk for relapse directly correlates with MRD level, that is, those with the highest MRD levels at the end of remission induction have the worst prognosis. High levels of MRD detected soon after beginning with postinduction therapy (weeks 12 to 14 of treatment) have also been associated with an increased risk for relapse, as have high levels measured in the peripheral blood as early as day 8 and in the marrow at day 15 of induction therapy.
Other Prognostic Factors
Gender and Race.
Some investigators have reported that male patients fare worse than female patients. This observation had been attributed, in part, to the risk for testicular relapse, although higher relapse rates in males have been observed even when testicular relapse rates were low. Race may also influence outcome, with lower EFS rates reported for African-American, Hispanic, and Native American patients, even after adjustment for differences in prognostically significant presenting features. The reasons why patients of different race have varying responses to therapy on certain regimens have not been fully elucidated but may in part be related to differences in the biologic subtypes of ALL observed in different groups. For example, African-American children have a higher relative incidence of T-cell ALL and lower rates of high hyperdiploidy, a favorable genetic subtype of precursor B-cell ALL. Hispanic children also have a lower frequency of ETV6-RUNX1 –rearranged ALL and, in one study, were noted to have a higher incidence of ALL with rearrangements of CRLF2, a subtype associated with an inferior prognosis.
Pharmacogenomics.
Several studies have suggested that outcome may be affected by how rapidly and effectively an individual patient metabolizes certain chemotherapeutic agents. Polymorphisms in genes involved in chemotherapy drug metabolism have been associated with risk for relapse. More favorable long-term outcomes have been reported in patients with mutant thiopurine methyltransferase (TPMT) phenotypes involved in the metabolism of thioguanines, such as 6-mercaptopurine, although such patients may also be at higher risk for developing significant treatment-related toxicities, including low blood cell counts, infection, liver dysfunction, and second malignancies. Investigations of other genes, such as the glutathione S-transferase genes (encoding enzymes involved in the intracellular detoxification of various compounds) and the folate metabolism genes (e.g., thymidylate synthase, methylenetetrahydrofolate reductase, and methylenetetrahydrofolate dehydrogenase) have led to conflicting results, without clear consensus regarding the prognostic significance of various polymorphisms.
Treatment Adherence.
Compliance with taking oral chemotherapy during the maintenance phase has been shown to impact outcome. In one study of pediatric ALL patients, nonadherence to 6-mercaptopurine (as assessed by electronic monitoring device that measured cap opening of the medication bottle) was associated with an inferior outcome. Adolescents and Hispanic patients were at higher risk for noncompliance. After adjusting for sex, NCI risk group, cytogenetics, ethnicity, and other factors, 6-mercaptopurine compliance was an independent prognostic factor, with a progressive increase in risk for relapse with decreasing levels of adherence.
Therapy
Historical Background
Over the past 50 years, there has been a dramatic improvement in the prognosis of children with ALL ( Fig. 50-3 ). Prior to 1947, when the first complete remission in childhood ALL was attained by Farber and colleagues, the median duration of survival from the time of diagnosis was 2 months. During the 1950s, drugs such as 6-mercaptopurine, methotrexate, and corticosteroids were found to be active in leukemia-bearing mice and subsequently in human leukemias. The first controlled clinical trials were conducted by Frei and associates, who ushered in the era of single-agent (and soon thereafter combination-agent) antileukemic chemotherapy trials. Active drugs introduced in the 1960s and 1970s included the anthracyclines (doxorubicin and daunorubicin), l -asparaginase, and the epipodophyllotoxins (etoposide and teniposide).
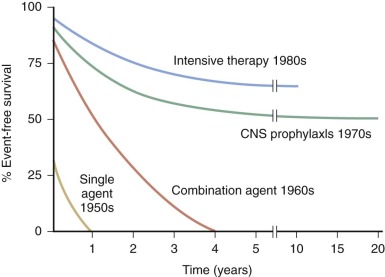
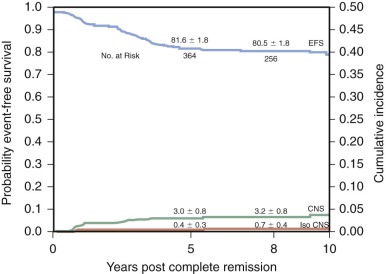
With current regimens for the treatment of childhood ALL, more than 95% of patients achieve complete remission and approximately 80% are long-term event-free survivors ( Table 50-3 and Fig. 50-4 ). Overall survival, which includes patients who are salvaged after relapse, is now approximately 90%. Improvement in outcome over the past 30 to 40 years occurred despite the fact that, with few exceptions, the drugs used for the treatment of ALL today were all available by the late 1960s and early 1970s. Improvement in cure rates can be attributed to many factors, including the development of complex chemotherapeutic regimens designed to achieve clonal eradication, improvements in supportive care, recognition of the CNS as a sanctuary site, and application of risk-directed therapy.
| Presenting Feature | No. of Patients | 10-Year EFS (±SE) | 10-Year OS (±SE) |
|---|---|---|---|
| Overall | 491 | 79 ± 2 | 89 ± 2 |
| Age at diagnosis (yr) | |||
| <1 | 14 | 42 ± 13 | 42 ± 13 |
| 1-9 | 385 | 82 ± 2 | 93 ± 1 |
| ≥10 | 92 | 75 ± 5 | 78 ± 4 |
| WBC (× 10 9 /L) | |||
| <10 | 239 | 83 ± 3 | 93 ± 2 |
| 10-49 | 155 | 80 ± 3 | 89 ± 3 |
| 50-99 | 43 | 70 ± 10 | 88 ± 5 |
| ≥100 | 54 | 66 ± 7 | 73 ± 6 |
| Immunophenotype | |||
| B-precursor cell | 438 | 78 ± 2 | 89 ± 5 |
| T-cell | 52 | 85 ± 5 | 90 ± 4 |
| Gender | |||
| Male | 274 | 78 ± 3 | 88 ± 2 |
| Female | 217 | 81 ± 3 | 91 ± 2 |
| B-Precursor cell National Cancer Institute (NCI) risk group | |||
| Standard | 303 | 83 ± 2 | 94 ± 2 |
| High | 121 | 72 ± 5 | 82 ± 4 |
Risk-Adapted Therapy
After the addition of CNS-directed therapy improved cure rates to approximately 50% in the 1970s, investigators compared presenting features in patients who were cured and those who relapsed to establish clinically relevant prognostic factors. Subsequent clinical trials used these prognostic factors to stratify therapy. More intensive therapy was administered to those patients considered to be at the highest risk for relapse. In contrast, some of the more morbid components of therapy were modified or eliminated for those children considered to have the best prognosis. The goal of risk-adapted therapy is to treat away adverse presenting features so that high-risk and low-risk patients have similar cure rates. For example, on Dana-Farber Cancer Institute (DFCI) ALL Consortium protocols, children with T-cell ALL (historically, a subgroup with an inferior prognosis) are treated as high-risk patients (receiving higher cumulative dosages of anthracycline and corticosteroid than lower-risk patients); with that therapy, their outcomes are as favorable as those with B-precursor cell disease.
For many years, pediatric cooperative groups and institutions applied prognostic factors differently when defining risk categories for clinical trials. A more uniform approach to risk classification was proposed and agreed on at an NCI-sponsored workshop held in 1993. For patients with B-precursor cell ALL, the standard-risk category was defined as age between 12 months and younger than 10 years and initial leukocyte count lower than 50,000/mm 3 . The remaining patients were considered to have high-risk ALL. Other characteristics used by the various cooperative groups to classify patients as high risk include T-cell phenotype, CNS-3 status at diagnosis, and slow early response to initial therapy, including a high peripheral blood absolute blast count at the end of a 7-day prednisone prophase. These various prognostic factors are discussed earlier.
Contemporary regimens have incorporated cytogenetic abnormalities and MRD levels into risk group stratification, and in some cases, only these factors are considered (and not more standard factors, such as age and leukocyte count) when assigning risk groups. Patients with certain chromosomal abnormalities (e.g., MLL gene rearrangements and low hypodiploidy) are treated as high risk, regardless of other presenting features. Similarly, end-induction MRD is used by almost all groups as a factor determining the intensity of postinduction treatment, with patients found to have higher levels allocated to more intensive therapies. On some protocols, MRD levels at a later time point (week 12) are also considered when risk-stratifying patients, because these have been shown to be prognostically important, especially in patients with T-cell ALL.
MRD measurements, in conjunction with cytogenetics, have also been used to identify subsets of patients with an extremely low risk for relapse who may be successfully treated with less intensive therapies. The Children’s Oncology Group reported a very favorable prognosis for patients with the B-precursor cell phenotype, NCI standard risk age/leukocyte count, CNS-1 status, and favorable cytogenetic abnormalities (either high hyperdiploidy with favorable trisomies or the ETV6-RUNX1 fusion) who had low peripheral blood MRD levels at day 8 and low marrow MRD at end of induction.
Ultimately, treatment is the most important prognostic factor, and those factors used to risk-stratify patients on one regimen may not be useful for patients treated on a different regimen.
Phases of Therapy
In general, treatment regimens for children with newly diagnosed ALL include three phases: remission induction, consolidation (or intensification), and continuation (or maintenance). The remission induction phase of therapy is designed to destroy measurable leukemia cells rapidly and minimize residual leukemia burden (i.e., the total number of leukemic cells in the body). The consolidation or intensification phase(s) is designed to reduce the total-body leukemia cell burden further and address issues of antileukemic drug resistance. The continuation or maintenance phase, consisting of low-dose chemotherapy, is designed to eradicate the residual leukemia cell burden. Throughout all phases, CNS-directed treatments (e.g., intrathecal chemotherapy) are an essential component because many systemically administered agents do not adequately penetrate the brain and spinal cord.
Remission Induction
After the diagnosis of ALL has been established and any urgent medical issues, such as metabolic imbalances, have been addressed, antileukemic chemotherapy should be initiated without delay. The initial phase of treatment, remission induction, is designed to reduce the leukemic cell burden to a clinically and hematologically undetectable level. Hematologic remission is defined as attainment of a normocellular bone marrow with 5% or fewer blasts and peripheral blood without lymphoblasts, with return of “normal” peripheral blood counts, that is, a granulocyte count exceeding 500 to 1000/mm 3 and a platelet count exceeding 100,000/mm 3 . Complete remission is defined as achievement of these criteria along with the absence of any signs and symptoms of extramedullary leukemia. A complete remission must be induced before the next component of therapy is begun. The duration of most induction regimens is 4 to 6 weeks.
Clinical trials in the 1960s demonstrated that combinations of two agents were consistently superior to single agents for inducing complete remission in children with ALL. Vincristine and prednisone produced complete remission in approximately 90% of pediatric patients with ALL. Addition of a third drug increased both the number of patients who achieved complete remission and the long-term relapse-free survival. Current induction regimens, therefore, consist of at least three agents, most often l -asparaginase or an anthracycline, in addition to vincristine and prednisone.
Many groups have further intensified induction regimens to include four or more agents. In theory, these more intensive induction regimens may prevent the emergence of drug-resistant leukemic clones by an initial leukemic cell lysis of greater rapidity and magnitude. The benefit in terms of long-term EFS of using four drugs during induction therapy (vincristine, prednisone, l -asparaginase, and an anthracycline) is widely accepted in higher-risk patients but less clear in lower-risk patients. Intensive induction regimens may enhance long-term EFS but can result in increased short-term morbidity, especially from infections during periods of granulocytopenia. However, even with four-drug induction regimens, few toxic deaths are reported (i.e., 1% to 2% of patients).
The initiation of CNS treatment is an integral component of induction therapy. After the diagnosis of ALL has been established by bone marrow examination, a lumbar puncture is performed for diagnostic and therapeutic purposes. It is generally recommended that intrathecal chemotherapy be given with the first diagnostic lumbar puncture to reduce the theoretical risk for inadvertently seeding the meninges with peripheral blood lymphoblasts. In addition to the diagnostic lumbar puncture, intrathecal chemotherapy is usually administered at least one more time during the first month of treatment. Patients with lymphoblasts present on their initial CSF specimen (CNS-2 or CNS-3 status, or traumatic lumbar puncture with blasts) typically receive more doses of intrathecal chemotherapy during the induction phase than those without any detectable CSF lymphoblasts (CNS-1 status).
Failure to achieve remission after 1 month of therapy (induction failure) is uncommon, occurring in fewer than 5% of patients. Induction failure is observed more commonly in patients with high presenting leukocyte counts or T-cell phenotype. Successful treatment of refractory ALL has been reported with the use of drugs such as cytarabine, epipodophyllotoxins (e.g., etoposide, teniposide), and anthracyclines (e.g., idarubicin). Nelarabine has been shown to induce remissions in patients with refractory T-cell ALL. Ultimately, 80% to 90% of children with initially refractory disease are able to achieve a complete remission. Despite this relatively high complete remission rate, overall survival for patients with a history of initial induction failure is poor (long-term overall survival rates of 20% to 30%). Allogeneic HSCT in first remission may improve the outcome for patients with initial induction failure. In a large retrospective series, a trend for superior outcome with allogeneic HSCT compared with chemotherapy alone was observed in patients with initial induction failure and either the T-cell phenotype (any age) or B-precursor cell phenotype and age younger than 6 years. In that study, B-precursor cell ALL patients who were aged 1 to 5 years at diagnosis and did not have any adverse cytogenetic abnormalities ( MLL translocation, BCR-ABL ) had a relatively favorable prognosis, without any advantage in outcome with the utilization of HSCT compared with chemotherapy alone.
Consolidation (Intensification) Therapy
The goals of consolidation therapy are to reduce the disease burden further and to adjust the intensity of treatment based on the risk for subsequent relapse. A wide variety of agents and schedules have been used during postinduction consolidation in pediatric trials. In general, there has been an attempt in lower-risk patients to limit exposure to agents associated with significant acute and late toxicities while reserving the use of more intensive therapies for patients at higher risk for relapse.
A commonly used postinduction consolidation regimen was first introduced by the German BFM study group. This consolidation scheme includes (1) a 4-week chemotherapy course (sometimes referred to as “consolidation” or “induction IB”) immediately after the initial induction phase consisting of cyclophosphamide, low-dose cytarabine, and a thiopurine (mercaptopurine or thioguanine), followed by (2) an interim maintenance phase, which includes multiple doses of either high-dose or escalating doses of methotrexate with or without leucovorin rescue, and then (3) a reinduction course (or delayed intensification), which typically includes the same agents used during the initial induction/consolidation cycles. Augmentation of this regimen, including the use of two delayed intensification cycles instead of one, has led to improved outcomes for higher-risk patients, including those with a slow early morphologic response to initial induction therapy. The DFCI ALL Consortium uses an alternative postinduction consolidation regimen that includes 20 to 30 weeks of l -asparaginase and, for higher-risk patients, additional doses of doxorubicin.
Continuation Therapy
Almost all current treatment regimens for ALL include a phase of continuation or maintenance therapy, in which patients are treated with less-intensive chemotherapy to complete at least 2 years of therapy. Most continuation regimens consist of weekly low-dose methotrexate and daily oral 6-mercaptopurine. Some groups have added regular pulses of vincristine and corticosteroid to this regimen, although their benefit remains controversial.
Several pharmacologic studies have documented highly variable bioavailability after oral administration of 6-mercaptopurine and methotrexate during the continuation phase. This may have important prognostic implications, because lower intracellular levels of thioguanine nucleotides and methotrexate polyglutamates, the major metabolites of 6-mercaptopurine and methotrexate, have been correlated with a higher risk for relapse. Variable bioavailability may be the result of various factors, including concurrent food, which may interfere with drug absorption. Bioavailability of oral mercaptopurine appears to be improved if it is given in the evening, without food or milk. Polymorphisms within genes involved in drug metabolism also affect rates of drug activation and clearance. For example, patients with deficiency in the activity of TPMT, an enzyme that inactivates mercaptopurine, have higher rates of toxicity during continuation therapy (but also may have better survival) than patients with normal TPMT activity. Most physicians attempt to compensate for the interindividual variations of bioavailability and metabolism by tailoring the dose of both drugs to the leukocyte count. Administration of maximally tolerated doses of methotrexate and 6-mercaptopurine during continuation therapy may improve outcome, as suggested by studies demonstrating that patients with lower leukocyte or neutrophil counts during continuation chemotherapy have lower relapse rates. Lack of patient compliance with oral medications, as well as physician compliance with protocol dosages, may adversely affect the efficacy of continuation chemotherapy.
6-Thioguanine is an alternative thiopurine that, unlike 6-mercaptopurine, does not rely on TPMT for its metabolism, resulting in higher intracellular levels of thioguanine nucleotides. 6-Thioguanine also has been shown to be more potent than 6-mercaptopurine in inducing cell death of lymphoblasts in vitro. The results of one randomized clinical trial comparing the two thioguanines indicated that 6-thioguanine was associated with a superior EFS (but not overall survival) in boys, primarily owing to a reduction in the frequency of CNS relapses. However, other randomized trials comparing the two thiopurines did not confirm an EFS advantage for 6-thioguranine. Additionally, the use of 6-thioguanine during the maintenance phase has been associated with significant hepatotoxicity, including veno-occlusive disease and cirrhosis, as well as higher remission death rates, primarily caused by infection. Thus 6-mercaptopurine remains the thiopurine of choice during the continuation phase.
Several investigators have studied the relative efficacy and toxicity of two corticosteroids used during postinduction therapy: prednisone and dexamethasone. Interest in substituting dexamethasone for prednisone stems from studies suggesting that dexamethasone has more potent in vitro antileukemic activity, higher free plasma levels, and enhanced CSF penetration. In several clinical trials conducted in the 1990s, the nonrandomized substitution of dexamethasone for prednisone appeared to affect outcome favorably. This finding was confirmed in several randomized clinical trials that indicated that dexamethasone was associated with a superior 5-year EFS. However, dexamethasone may also be associated with increased corticosteroid-related toxicities, including higher rates of fracture, osteonecrosis, and infectious complications, especially in older children and adolescents. Thus the optimal corticosteroid preparation and dosing have yet to be determined and may differ based on presenting features (e.g., age) and risk for relapse.
Attempts to intensify continuation therapy by administering rotating pairs of non–cross-resistant drugs, including many agents not traditionally incorporated during the continuation phase, such as cyclophosphamide, epipodophyllotoxins, and cytarabine, have led to mixed results. In studies conducted by St. Jude Children’s Research Hospital, this strategy resulted in improved outcomes for higher-risk but not lower-risk patients but also was associated with high rates of toxicity, including secondary AML. Based on this risk, these agents (especially epipodophyllotoxins) are, in general, no longer administered to most patients during the continuation phase.
CNS-Directed Therapies
In the 1960s, as systemic chemotherapy became more effective in prolonging the duration of hematologic remission in patients with ALL, the incidence of CNS leukemia as an initial site of relapse became progressively more common. It was hypothesized that leukemia cells, even if subclinical, were present in the CNS in all patients and that these cells were protected by the blood-brain barrier from systemically administered chemotherapy. Thus, the concept of the CNS as a “pharmacologic sanctuary” (i.e., an anatomic space that is poorly penetrated by systemically administered chemotherapeutic agents) emerged. The introduction of radiation therapy to treat subclinical CNS leukemia in the 1970s was a pivotal step in boosting long-term disease-free survival rates in childhood ALL to 50%. Although most pediatric patients are now treated without prophylactic cranial radiation, the importance of effectively treating subclinical CNS leukemia is undisputed for childhood ALL.
All successful treatment regimens for ALL include therapy directed at treating CNS leukemia. Options for CNS-directed therapies include cranial irradiation, intrathecal chemotherapy, and CNS penetrant systemic chemotherapy.
Radiation therapy was the first modality successfully used to prevent CNS relapses. The use of radiation for CNS treatment was based on experiments demonstrating that L1210 murine leukemia could be cured when cranial irradiation was added to systemic treatment with cyclophosphamide. In the 1960s and 1970s, studies performed at St. Jude Children’s Research Hospital documented the effectiveness of CNS radiation as preventive therapy in children with ALL. In one study, patients were randomized to receive 2400 cGy of craniospinal radiation or no radiation. The difference in CNS relapse rates was striking: only 2 of 45 irradiated patients (4%) had their initial complete remissions ended by meningeal relapse, compared with 33 of 49 nonirradiated patients (67%). Moreover, 31 of the patients who received prophylactic radiation remained alive after 18 to 20 years, compared with only 10 patients in the nonirradiated group. Subsequent studies demonstrated that 2400-cGy cranial irradiation with intrathecal methotrexate was as effective in preventing CNS relapse as 2400-cGy craniospinal irradiation without intrathecal chemotherapy. Because craniospinal radiation was associated with increased toxicity, including excessive myelosuppression and spinal growth retardation, radiation directed only to the cranium administered with intrathecal chemotherapy became the standard form of CNS treatment in the 1970s.
Although 2400-cGy cranial irradiation (with intrathecal chemotherapy) effectively prevents CNS relapses in children with ALL, it is also associated with subsequent learning disabilities, growth and neuroendocrinologic abnormalities, and an increased risk for second malignant neoplasms. Thus many investigators have studied alternative CNS treatments, including lower doses of cranial radiation (1200 to 1800 cGy), systemically administered CNS-penetrant chemotherapy (i.e., agents known to achieve therapeutic levels in CSF), or intrathecal chemotherapy. Current regimens for childhood ALL include some or all of these CNS-directed therapies.
The proportion of patients receiving cranial radiation has decreased significantly over the past several decades; on current regimens, most pediatric patients are treated without radiation. When it is administered, cranial radiation is generally restricted to patients with a higher risk for relapse involving the CNS, especially those with T-cell disease, high presenting leukocyte counts, or CNS-3 status at diagnosis. Doses lower than 2400 cGy have been shown to be effective, and therefore are typically used when cranial radiation is administered. For instance, CNS and marrow relapse rates were not significantly different when the dose of cranial radiation was lowered from 2400 to 1800 cGy in consecutive Children’s Cancer Study Group trials. The BFM and DFCI groups have successfully used 1200-cGy irradiation in the context of an intensive systemic regimen that included high-dose methotrexate (known to penetrate the CNS effectively). Irradiated patients also typically receive intermittent intrathecal chemotherapy in addition to cranial irradiation as CNS preventive therapy.
Systemic therapy plays an important role in the prevention of CNS leukemia. Penetration of the CSF by drugs has been clearly demonstrated with the use of glucocorticoids and high doses of methotrexate and cytarabine. It has also been shown that systemically administered asparaginase, whose efficacy is a function of asparagine depletion, effectively lowers CSF asparagine levels.
Intrathecal chemotherapy, used in conjunction with intensive systemic therapy and/or cranial irradiation, is an important component of CNS treatment. Many investigators have replaced cranial irradiation with frequent dosing of intrathecal chemotherapy, especially in lower-risk patients. The success of these efforts appears to depend on the frequency with which intrathecal chemotherapy is delivered, as well as the intensity and CNS penetration of the systemic chemotherapy administered. Investigators from the Children’s Cancer Group demonstrated that administering intrathecal methotrexate throughout all phases of therapy was as effective in preventing CNS relapses in lower-risk patients as 1800-cGy cranial irradiation combined with only 6 months of intrathecal methotrexate, but only in the context of an intensified systemic regimen. When a less-intensive systemic regimen was used, more CNS relapses were observed in nonirradiated patients. Similarly, in a DFCI ALL Consortium study, excessive CNS relapses were observed in standard-risk males when cranial irradiation was eliminated without increasing the frequency of intrathecal chemotherapy or the intensity of systemic chemotherapy. However, in a subsequent study conducted by the same group, standard-risk patients who received more frequent doses of intrathecal chemotherapy had as low a CNS relapse rate as irradiated patients. Some controversy exists regarding the relative efficacy of intrathecal methotrexate alone and “triple intrathecal chemotherapy” (intrathecal methotrexate, cytarabine, and hydrocortisone). In one randomized study in standard-risk patients, triple intrathecal chemotherapy was associated with a lower rate of isolated CNS relapse but there was no difference in overall EFS. Randomized studies have also demonstrated that the use of dexamethasone instead of prednisone, which is believed to be less CNS penetrant, is associated with a lower rate of isolated CNS relapses in nonirradiated patients. Thus those regimens that have successfully eliminated cranial radiation include extended and/or frequent dosing of intrathecal chemotherapy in conjunction with intensified, more CNS penetrant systemic therapy, including dexamethasone and/or high- or intermediate-dose methotrexate.
Several clinical trials have been conducted on which cranial radiation was omitted for all patients, including high-risk subsets. Most of these trials include multiple doses of either high-dose methotrexate or high-dose cytarabine and increased frequency of intrathecal chemotherapy. While the overall EFS on these trials was similar to that of contemporaneously conducted trials on which some patients received prophylactic radiation, some nonirradiated patient subsets had a significantly high risk for CNS relapse, including patients with T-cell phenotype or the presence of blasts in CSF at diagnosis. Additionally, some patients with high-risk features on these trials were treated with allogeneic SCT in first complete remission (which included total-body irradiation).
The goal of eliminating cranial radiation in pediatric patients is to minimize long-term sequelae (see later). Although several studies have indicated that intrathecal chemotherapy and intensive systemic therapy can be as effective as cranial irradiation in preventing CNS relapses, the relative late neurocognitive toxicities of these CNS treatment strategies remains unsettled. Moreover, higher rates of acute neurotoxicity, including seizures, have been observed with regimens that included multiple courses of high-dose methotrexate and frequent doses of intrathecal chemotherapy.
Cessation of Treatment
The optimal duration of therapy remains unknown. Most investigators continue to treat patients for 2 to 3 years, based on results of older studies, in which patients received therapy that was less intensive than in current regimens. Randomized studies conducted by the Children’s Cancer Group in the 1970s indicated that there was no significant difference in outcome when comparing 5 versus 3 years of therapy. In the Childhood Leukaemia Trial UKALL VIII, a randomized study of 2 versus 3 years of continuation therapy conducted in the 1980s, the researchers observed no significant survival advantage with longer therapy. Patients receiving the shorter duration of therapy had a higher relapse rate, but this was counterbalanced by a higher remission death rate in those receiving 3 years of treatment. Some early studies suggested that the optimal duration of therapy may be different for boys and girls, with boys benefiting from a more prolonged continuation phase, although this finding may be less relevant with current regimens.
Even with more intensive regimens, attempts to shorten therapy duration from 2 years have not been successful. The BFM group randomized patients to receive 18 or 24 months of treatment and observed a higher relapse rate with patients who received the shorter course of treatment. Similarly, high relapse rates were observed in a nonrandomized study conducted by the Tokyo Children’s Cancer Study Group, in which patients received intensified therapy for only 12 months, suggesting that truncated therapy, even if intensive, is inadequate for most children with ALL.
Treatment of Special Patient Subsets
Philadelphia Chromosome–Positive ALL Patients
The Ph chromosome, t(9;22)(q34;q11), which leads to fusion of the BCR and ABL genes, is detectable in approximately 5% of children and 20% of adults with ALL. In pediatric ALL, it is more frequent in adolescents than in younger children. When treated on regimens with conventional chemotherapeutic agents, these patients have a dismal prognosis, with long-term EFS rates ranging from 0% to 25%. Allogeneic SCT in first remission has been shown to lead to more favorable outcomes and, prior to the availability of tyrosine kinase inhibitors, was considered the treatment of choice for children with Ph chromosome–positive ALL. In one study, which reviewed the outcome of 610 children with Ph chromosome–positive ALL treated by 10 study groups in the pre–tyrosine kinase inhibitor era between 1995 and 2005, transplantation in first remission from an HLA-matched related or unrelated donor was associated with a higher likelihood of disease-free survival compared with chemotherapy alone.
Imatinib mesylate is a selective inhibitor of the BCR-ABL tyrosine kinase. Phase I and II studies of single-agent imatinib in children and adults with relapsed or refractory Ph chromosome–positive ALL have demonstrated relatively high response rates, although these responses tended to be of short duration. Results from pediatric trials have suggested that the administration of imatinib in combination with multiagent chemotherapy is feasible, without excessive toxicity. In a pilot trial conducted by the Children’s Oncology Group, patients treated with the combination of chemotherapy and imatinib (but without transplant) appeared to do at least as well as other patients on the same trial who underwent SCT in first remission.
Infants
Patients diagnosed during the first year of life represent 2% to 4% of cases of childhood ALL. The outcome of infants with ALL is significantly worse than that for older children, with reported long-term EFS rates of 20% to 40%. The major cause of treatment failure is relapse, which tends to occur early, often in the first 1 to 2 years after diagnosis. In vitro chemosensitivity assays have indicated that lymphoblasts from infants with ALL are relatively resistant to agents commonly used in childhood ALL regimens, such as corticosteroids and l -asparaginase. Infants also appear to be more vulnerable to the toxic effects of therapy, especially severe infections.
The poor outcome of infants appears to be related to the biologic distinctiveness of the disease observed in this age group. Up to 80% of infants with ALL present with molecularly detectable rearrangements of the MLL gene on chromosome 11q23. The presence of a MLL gene rearrangement is an independent predictor of poor outcome in infant ALL, with the worst outcomes observed in MLL -rearranged patients who present at a very young age (<6 months at diagnosis) and with high presenting leukocyte counts.
Because of their poor prognosis, infants with ALL are usually treated on separate protocols with intensified therapy. In vitro drug sensitivity testing and small, nonrandomized clinical trials have suggested that high-dose cytarabine, an agent more often used in AML, might benefit infants with ALL. In the Interfant-99 trial, the largest clinical trial conducted for infants with ALL, a cytarabine-intensive regimen resulted in a 4-year EFS of 47%.
The role of allogeneic SCT in first complete remission in infants with ALL remains controversial. On the Interfant-99 trial, a subset analysis suggested that SCT in first remission may be beneficial in infants with high-risk features ( MLL gene rearrangement, age younger than 6 months at diagnosis, and presenting leukocyte count at or above 300,000/µL); an EFS advantage for transplant was not demonstrated in other MLL -rearranged infants.
Adolescents
Older children and adolescents (aged 10 to 18 years at diagnosis) have a less favorable outcome than children aged 1 to 10 years at diagnosis, and more aggressive treatments are generally administered to these patients. Adolescents present more frequently with adverse prognostic features, such as a high leukocyte count, T-cell immunophenotype, and the BCR-ABL translocation, and a lower frequency of favorable features, such as high hyperdiploidy and the ETV6-RUNX1 fusion gene. Adolescents also appear to be at higher risk than younger children for developing treatment-related complications, including osteonecrosis, pancreatitis, and thrombosis. Several investigators have compared outcomes of older adolescents with ALL (aged 15 to 20 years) treated in pediatric or adult clinical trials, and all have found that these patients fare better with a pediatric regimen. The superior outcome of older adolescents treated with pediatric ALL regimens may be related, in part, to the components of therapy; pediatric regimens tend to include higher cumulative dosages of vincristine, corticosteroid, and l -asparaginase than treatments designed for adults with ALL.
Children with Down Syndrome
Children with Down syndrome have increased risk for developing both ALL and AML. Approximately one half to two thirds of cases of acute leukemia in children with Down syndrome are ALL. Although the vast majority of cases of AML in children with Down syndrome occur before the age of 4 years, ALL in children with Down syndrome has an age distribution similar to that of ALL in non–Down syndrome children. Patients with ALL and Down syndrome almost always present with B-precursor cell phenotype and have a lower incidence of favorable and unfavorable cytogenetic abnormalities. They appear to be at higher risk for developing treatment-related complications, especially those related to methotrexate, such as mucositis and myelosuppression. Outcomes for children with Down syndrome and ALL have generally been reported to be less favorable than those of non–Down syndrome patients, which may be related to a higher incidence of treatment-related mortality because of infection and a lower frequency of favorable biologic features, such as high hyperdiploidy and the ETV6-RUNX1 fusion.
High expression of CRLF2 occurs in 50% to 60% of cases of ALL in children with Down syndrome, compared with 5% to 10% of non–Down syndrome B-precursor cell ALL patients. A higher frequency of activating mutations in JAK2 has also been observed in Down syndrome ALL (~20% of cases) ; almost all patients with JAK2 mutations also have CRLF2 overexpression. It does not appear that CRLF2 genomic aberrations or JAK2 mutations have independent prognostic relevance in Down syndrome ALL. However, in one study, deletions of the IKZF1 gene, observed in 35% of Down syndrome ALL cases (and frequently associated with CRLF2 overexpression), was found to be a strong independent predictor of inferior EFS.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


