Study group
Trial
Years
ALL subtypes
No. of patients
Age
EFS, % (year)
OS, % (year)
Reference
AIEOP/BFM
AIEOP-BFM ALL 2000
2000–2006
B-ALL
4016
1–18
80.4 (7)
91.8 (7)
Conter et al. [1]
T-ALL
464
75.9 (7)
80.7 (7)
Schrappe et al. [2]
MRC
UKALL 2003
2003–2011
All
3126
1–24
87.2 (5)
91.5 (5)
Vora et al. [3]
B-ALL
2731
–
–
T-ALL
388
–
–
DCOG
DCOG Protocol ALL-9
1997–2004
All
859
1–18
81 (5)
86 (5)
Veerman et al. [4]
B-ALL
701
82 (5)
–
T-ALL
90
72 (5)
–
EORTC-CLG
EORTC CLG 58591
1998–2008
All
1947
1–18
82.7 (5)
89.7 (5)
Domenech et al. [5]
B-ALL
1650
–
–
T-ALL
296
–
–
NOPHO
ALL-2000
2002–2007
All
1023
1–15
79 (5)
89 (5)
Schmiegelow et al. [6]
B-ALL
906
–
–
T-ALL
115
–
–
COG
Various CCG, POG, and COG trials
2000–2005
All
7153
0–22
–
90.4 (5)
Hunger et al. [7]
B-ALL
5982
–
91.1 (5)
T-ALL
459
–
81.6 (5)
SJCRH
Total therapy XV
2000–2007
All
498
1–18
85.6 (5)
93.5 (5)
Pui et al. [8]
B-ALL
422
86.9 (5)
94.6 (5)
T-ALL
76
78.4 (5)
87.6 (5)
DFCI
DFCI 05-001
2005–2010
All
551
1–18
85 (5)
91 (5)
Place et al. [9]
B-ALL
482
85 (5)
91 (5)
T-ALL
69
87 (5)
91 (5)
Ma-Spore
Ma-Spore ALL 2003
2002–2011
All
556
0–18
80.6 (6)
88.4 (6)
Yeoh et al. [10]
B-ALL
507
80.7 (6)
–
T-ALL
49
80.5 (6)
–
TCCSG
TCCSG L99-15
1999–2003
All
754
1–18
78.2 (4)
87.6 (4)
Hasegawa et al. [11]
B-ALL
664
80.5 (4)
–
Manabe et al. [12]
T-ALL
90
66.0 (4)
–
2.2 Epidemiology
Registry of the Japanese Society of Pediatric Hematology (currently, the Japanese Society of Pediatric Hematology and Oncology) covers more than 90% of the population under age 20 with hematological malignancies in Japan. 2463 patients are diagnosed as ALL between 2006 and 2010, that is, approximately 500 new cases are diagnosed every year [13]. As annual number of childhood cancer in Japan is estimated as 2000–2500, ALL accounts for 20–25% of all childhood cancers and 72% of all childhood leukemias. Male/female ratio is 1.3:1, and the peak incidence of ALL occurs between 2 and 5 years old. Immunophenotypically, 86% is B-cell precursor, 11% is T cell, 2% is mature B cell, and 1% is unknown.
Several inherited syndromes are known to be associated with an increased risk of ALL. Among all, most well recognized is Down syndrome which is 10–20 times more likely to develop ALL compared to non-Down syndrome children [14]. In addition, several rare germline mutations, such as PAX5 and ETV6, have been identified to be a genetic predisposition to B-ALL in familial leukemia kindreds [15, 16]. Although it has been long recognized that majority of ALL patients have no apparent clinical inherited factors, recent genome-wide association studies have identified several germline single-nucleotide polymorphisms (SNPs) in several genes (including IKZF1, ARID5B, CEBPE, CDKN2A, GATA3, etc.) that are associated with significantly higher risk of developing ALL [17–20].
Environmental factors, such as ionizing radiation, electromagnetic field, certain diets (e.g., bioflavonoids), seem to have little association in most of the ALL cases [21–23]. However, two infection-based hypotheses, Greaves’ “delayed infection” hypothesis and Kinlen’s “population-mixing” hypothesis that childhood ALL arise as a consequence of an abnormal immune response in susceptible individuals to common infections, are well supported by epidemiological data [24, 25].
2.3 Pathobiology
It is considered that ALL occurs as a consequence of malignant transformation of an abnormal single lymphoid progenitor cell via multi-step genetic alterations. It is not entirely clear how and when these genetic events take place. However, it is believed that causation of ALL is by chance, and normal allelic variation in inherited genes or inherited ALL predisposing syndromes in a small portion of cases, and exposures to various exogenous and endogenous factors all contribute to leukemogenesis [26].
Identification of genetic abnormalities plays a pivotal role in understanding the biology and treatment of ALL. Cytogenetic analysis by karyotyping which was established in 1960s and by fluorescence in situ hybridization (FISH) in 1980s is still necessary in the modern diagnostic evaluation in ALL. However, newer array-based technologies enabled us to analyze global gene expression, DNA copy numbers, SNPs, and methylation status. In addition, recent advance in genome-wide sequencing which include whole-genome sequencing, transcriptome sequencing (RNA-seq), and whole-exome sequencing have helped us to approach the true nature of ALL. Although these novel genomic techniques are still used as research discovery tools, it is expected that they would be increasingly utilized for clinical applications in the near future.
Recurrent genetic alterations in ALL include aneuploidy (changes in chromosome number) and structural abnormalities. The molecular and clinical features of specific alterations are discussed below (Fig. 2.1).
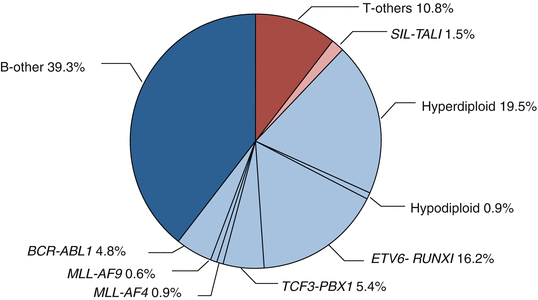
Fig. 2.1
Cytogenetic and molecular genetic abnormalities in pediatric ALL. (data provided from the TCCSG L04-16 and L07-16 studies)
2.3.1 Aneuploidy
Ploidy (number of chromosomes of a cell) could be determined by direct counting of the chromosome numbers in a metaphase karyotype preparation by G-banding technique. Clinically, high hyperdiploidy is defined as chromosome number greater than 50 and hypodiploidy as less than 45 chromosomes. DNA index (DI) is an alternative method of measuring DNA content by flow cytometry; DI is 1.0 in normal diploid cells, while it is 1.16 or higher in high hyperdiploid cases.
2.3.1.1 Hyperdiploidy
High hyperdiploidy accounts for 20–30% of childhood ALL. There is a distinct pattern of chromosomes gained at each modal number of chromosomes: most commonly gained is chromosome 21 (ch21), followed by ch4, chX, ch10, ch6, ch14, ch18, and ch17 [27]. Previous clinical studies have demonstrated that certain combination of gained chromosome are associated with better prognosis: “double trisomy” of ch4 and ch10 in the Pediatric Oncology Group (POG); “triple trisomy” of ch4, ch10, and ch17 in Children’s Oncology Group (COG); trisomy of ch11 and ch17 in the Tokyo Children’s Cancer Study Group (TCCSG) [27–29]. Although prognostic significance of specific gained chromosomal combination is inconsistent among different study groups, high hyperdiploidy itself is associated with CD10-positive B-cell precursor phenotype, low leukocyte (WBC) count, younger age at diagnosis, and excellent prognosis.
2.3.1.2 Hypodiploidy
In contrast, hypodiploidy accounts for only 1% of childhood ALL and is a strong predictor of poor prognosis [30, 31]. Recent genomic analysis has identified TP53 mutations in 91% of low-hypodiploid ALL (32–39 chromosomes) [32]. Surprisingly, the mutation was also present in germline in 43% of the cases, which is a hallmark of Li-Fraumeni syndrome. Occasionally, there are cases with “masked hypodiploidy,” which occurs as a result of doubling of a hypodiploid clone [33]. Caution is needed because they do not appear to be different from non-masked hypodiploidy in clinical and prognostic features but could be misdiagnosed as “hyperdiploidy” harboring opposite prognosis.
2.3.2 Structural Chromosomal Abnormalities
Structural chromosomal abnormalities, most commonly translocations, are frequently observed in childhood ALL. They are considered to be initiating events in leukemogenesis. Several occur in utero, nearly 100% of MLL-AF4, similar to hyperdiploid cases, and 75% of ETV6-RUNX1, but none of TCF3-PBX1 [34]. This is supported by evidences of the high rate of concordance of leukemia in monozygotic twins and the prenatal origin of ALL demonstrated directly by the detection of unique fusion in neonatal blood spots on Guthrie cards who later developed ALL [35, 36]. Generally, two functional classes of translocations exist. One is juxtaposition of oncogenes with regulatory regions of actively transcribed genes causing its dysregulated target gene expression, such as c-MYC to immunoglobulin gene in Burkitt’s lymphoma/leukemia. Another is fusion of the genes at the translocation breakpoints to encode a novel chimeric protein with oncogenic function, ETV6-RUNX1, TCF3-PBX1, BCR-ABL1, rearrangements of mixed-lineage leukemia (MLL or KMT2A) gene, etc.
2.3.2.1 ETV6-RUNX1 (TEL-AML1)
t(12;21)(p13;q22.1) accounts for 15–20% of childhood ALL. It is cryptic in most cases and could be detected by FISH or real-time quantitative polymerase chain reaction (PCR). The translocation results in fusion of two hematopoietic transcription factor genes, ETV6 (formerly known as TEL) and RUNX1 (AML1). ETV6-RUNX1 appears to arise in utero, but the fusion solely itself is not sufficient to cause the leukemia and subsequent events are required to full progression [37]. Children with ETV6-RUNX1 ALL is associated with excellent outcome similar to high hyperdiploid ALL [38].
2.3.2.2 TCF3-PBX1 (E2A-PBX1)
t(1;19)(q23;p13.3) is the second most common translocation and accounts for 5% of childhood ALL. The translocation results in the fusion of two transcription factor genes, TCF3 (formerly known as E2A) and PBX1. TCF3-PBX1 ALL appears to be pre-B cell phenotype with positive cytoplasmic μ. It was once associated with poor prognosis but has lost its prognostic significance in the context of modern ALL chemotherapy especially that contains high-dose methotrexate [39]. However, TCF3-PBX1 is associated with higher risk of central nervous system (CNS) relapse [40].
There is a rare subtype of TCF3 gene involved ALL with fusion partner hepatic leukemia factor (HLF) gene which arise from t(17;19)(q22;p13). TCF3-HLF only occurs in less than 1% of childhood ALL but has unique characteristics such as hypercalcemia, an increased risk of disseminated intravascular coagulation (DIC), and, moreover, very poor prognosis [41].
2.3.2.3 KMT2A (MLL) Gene Rearrangements
KMT2A (MLL) gene, located at chromosome band 11q23, encodes a histone methyltransferase that is involved in epigenetic regulation of blood cell development via expression of multiple Hox genes [42]. Rearrangements of MLL occur as a result of balanced chromosomal translocations that fuse the MLL gene to one of more than 70 known partner genes. The most common in ALL is MLL-AF4 (KMT2A-AFF1) derived from t(4;11)(q21;q23), followed by MLL-ENL (KMT2A-MLLT1) from t(11:19)(q23;p13), and MLL-AF9 (KMT2A-MLLT3) from t(9;11)(p22;q23) [43]. MLL rearrangement is particularly common in ALL in infants (age <1 year old), which accounts for nearly 80% of the cases and associated with CD10-negative pro-B cell phenotype, high leukocyte count at diagnosis, and very poor prognosis (Table 2.2) [44–48]. ALL with MLL rearrangements have very few additional somatic mutations, one of the lowest of any sequenced cancers [49].
Table 2.2
Results of the recently reported trials for infants with ALL
Study group | Trial | Years | ALL subtypes | No. of patients | No. of patients received Allo-SCT in 1CR | EFS, % (year) | OS, % (year) | Reference |
|---|---|---|---|---|---|---|---|---|
Interfant | Interfant-99 | 1999–2005 | All | 482 | 37 | 47.0 (4) | 55.3 (4) | Pieters et al. [44] |
MLL-r | 314 | 36.9 (4) | – | |||||
MLL-g | 82 | 74.1 (4) | – | |||||
CCG | CCG 1953 | 1996–2000 | All | 115 | 37 | 41.7 (5) | 44.8 (5) | Hilden et al. [45] |
MLL-r | 79 | 33.6 (5) | – | |||||
MLL-g | 36 | 60.3 (5) | – | |||||
COG | COG P9407 (cohort 3) | 2001–2006 | All | 147 | 0 | 42.3 (5) | 52.9 (5) | Dreyer et al. [46] |
MLL-r | 100 | 35.5 (5) | – | |||||
MLL-g | 35 | 69.7 (5) | – | |||||
JILSG | MLL96 and MLL98 | 1995–2001 | All | 102 | 49 | 50.9 (5) | 60.5 (5) | Tomizawa et al. [47] |
MLL-r | 80 | 38.6 (5) | 50.8 (5) | |||||
MLL-g | 22 | 95.5 (5) | 95.5 (5) | |||||
JPLSG | MLL03 | 2004–2009 | MLL-r | 62 | 44 | 43.2 (4) | 67.2 (4) | Koh et al. [48] |
2.3.2.4 BCR-ABL1
t(9;22)(q34;q11.2) results in formation of Philadelphia (Ph) chromosome, which is one of the first leukemic translocations described. Ph encodes BCR-ABL1, an activated tyrosine kinase. It is the most common translocation in adult ALL (25% of the cases) but less common in children (less than 5% of the cases). Ph-positive (Ph+) ALL was one of the most difficult to cure ALL with only 30–40% event-free survival (EFS) rate despite intensive chemotherapy and allogeneic hematopoietic stem cell transplantation [50]. However, introduction of imatinib, a selective tyrosine kinase inhibitor (TKI), has revolutionized the therapy for Ph+ ALL (Table 2.3) [51–54].
Table 2.3
Results of the recently reported trials for children with Philadelphia chromosome positive ALL in the TKI era
Study group | Trial | Years | Subgroups | No. of patients | No. of patients received Allo-SCT in 1CR | DFS, % (year) | OS, % (year) | Reference |
|---|---|---|---|---|---|---|---|---|
EsPhALL | EsPhALL | 2004–2009 | All | 178 | 137 | 61.9 (4) | 72.1 (4) | Biondi et al. [51] |
Good-risk with imatinib | 46 | 37 | 72.9 (4) | – | ||||
Good-risk without imatinib | 44 | 32 | 61.7 (4) | – | ||||
Poor-risk | 70 | 59 | 53.5 (4) | 63.5 (4) | ||||
COG | AALL0031 (cohort 5) | 2002–2006 | Chemotherapy + imatinib | 28 | 0 | 70 (5) | – | Schultz et al. [52] |
Sibling donor BMT | 21 | 21 | 65 (5) | – | ||||
Unrelated donor BMT | 13 | 13 | 59 (5) | – | ||||
JPLSG | ALL-Ph04 | 2004–2008 | All | 42 | 26 | 54.1a (4) | 78.1 (4) | Manabe et al. [53] |
2.3.2.5 Philadelphia (Ph)-like ALL
Ph-like or BCR-ABL1-like ALL is a newly recognized entity, harboring similar gene expression profile to Ph+ ALL but without BCR-ABL1 fusion gene [55, 56]. Recent genomic analyses have revealed diverse range of genetic alterations that activate tyrosine kinase signaling in 90% of the cases. The most commonly identified alterations are rearrangements involving ABL1, ABL2, CRLF2, CSF1R, EPOR, JAK2, NTRK3, PDGFRB, PTK2B, TSLP, or TYK2 and sequence mutations involving FLT3, IL7R, or SH2B3 [57]. Importantly, “ABL-class” kinases (ABL1, ABL2, CSF1R, and PDGFRβ) could be targeted with TKI such as imatinib and dasatinib and alterations that activate JAK-STAT signaling (JAK1, JAK2, JAK3, CRLF2, EPOR, TSLP, and IL7R) with JAK inhibitors [58].
2.3.2.6 iAMP21
ALL with intrachromosomal amplification of chromosome 21 (iAMP21) is characterized by amplification of a portion of ch21, which could be detected by FISH analysis using a probe for the RUNX1 gene that reveals five or more copies of the gene (or three extra copies on a single abnormal ch21 in metaphase FISH) [59]. It occurs in 2% of childhood ALL and is associated with poor prognosis [60].
2.3.2.7 IKZF1, CRLF2, and JAK
Except for MLL-rearranged ALL in infants, many of the subtypes have multiple additional genetic alterations in general. These alterations commonly target genes encoding proteins involved in cell signaling, tumor-suppression functions, and lymphoid differentiation.
Most commonly targeted genes involved in B-lymphoid development are PAX5 and IKZF1 that are mutated in 31% and 15% of children with B-ALL, respectively, [61]. Notably, recurrent deletions and inactivating mutations in IKZF1, which encodes the hematopoietic transcription factor IKAROS, are associated with very poor prognosis in B-ALL [56]. Genetic alterations of IKZF1 occur more frequently in high-risk cases, including Ph+ ALL and Ph-like ALL [62].
Cytokine receptor-like factor 2 (CRLF2) forms a heterodimeric cytokine receptor with interleukin-7 receptor α (IL7Rα) that mediates B-cell precursor proliferation and survival via activation of downstream JAK/STAT pathways. Rearrangements of CRLF2 gene as IGHa-CRLF2 or P2RY8-CRF2 result in CRLF2 overexpression and found in 5–8% of pediatric ALL, 10–15% of adult ALL, and more than 50% of Down syndrome ALL [63–65]. It has been reported as poor prognostic factor but still controversial.
2.3.2.8 ETP-ALL
In contrast to genetic alterations discovered in B-ALL, many of the genetic alterations in T-ALL have not been found to have prognostic value yet. However, one subset of T-ALL with unique biology, early T-cell precursor (ETP) ALL that accounts for 10–15% of pediatric T-ALL, is recognized as a new entity [68]. ETP-ALL was originally identified by its unique gene expression pattern and immunophenotype with very early T-cell progenitor features: absence of T-cell markers CD1a and CD8; dim or absent CD5; combined with cytoplasmic, but not surface, CD3; and positive for one or more myeloid/stem cell markers (CD34, CD117, HLA-DR, CD13, CD33, CD11b, or CD65). By whole-genome sequencing, gene mutations similar to myeloid leukemias including RAS signal pathways were identified [69].
2.3.2.9 Genetic Alterations at Relapse
Genomic studies of matched diagnosis and relapsed ALL sample pairs have revealed that only 42% of the cases had evolved from diagnosis clone (8% was same and 34% was clonal evolution from diagnosis clone), but majority (52%) was rather evolved from ancestral clones, and the remaining 6% was genetically distinct secondary leukemia [70]. By further sequencing studies, mutations including CREBBP, TP53, and NT5C2 were identified [71–74]. Focal deletion and sequence mutations in CREBBP were found in 19% of children with relapsed B-ALL. The mutation causes impaired histone acetylation and transcriptional regulation of CREBBP targets, thus impairing the normal CREBBP-mediated transcriptional response to glucocorticoids and possibly results in steroid therapy resistance of the relapsed clone. NT5C2 encodes as 5′-nucleotidase enzyme that is involved in metabolisms of 6-mercaputopurine. Its mutation causes increased enzyme activity and resistance to nucleoside analog therapy. TP53 alterations were identified in 12% of relapsed B-ALL and 6% of relapsed T-ALL cases in the Berlin-Frankfurt-Münster (BFM) study, which was enriched compared to the initial diagnosis samples and was associated with poor response to the therapy and inferior outcome.
2.4 Clinical Management
2.4.1 Clinical Presentation
Anemia, thrombocytopenia, and/or neutropenia are typically observed among ALL patients, which reflect failure of normal hematopoiesis; pallor, fatigue, bleeding (e.g., petechiae or purpura), and fever (usually as leukemia-related rather than infection) are often present. Hepatosplenomegaly, lymphadenopathy, and bone pain are frequently manifested. Although not common at the time of initial diagnosis, involvement of extramedullary sites such as CNS, testis, and skin might be present simultaneously. Duration of these symptoms may vary from days to months; however, one may face life-threatening “oncologic emergency” situation at initial presentation, superior vena cava syndrome/superior mediastinal syndrome in T-ALL patients, disseminated intravascular coagulation, tumor lysis syndrome (renal failure, elevated levels of serum uric acid, potassium, and/or phosphate), and CNS hemorrhage or thrombosis induced by hyperviscosity syndrome, both often associated with hyperleukocytosis (>100,000/μL in peripheral blood), etc.
2.4.2 Diagnostic Procedures
Diagnosis of ALL is established by morphological detection of more than 25% leukemic blasts in bone marrow by aspirated bone marrow smears. It is preferable to perform bone marrow biopsy in case of dry tap. In addition to morphological examination, immunophenotyping, karyotype, and genetic analyses are essential for ALL diagnosis.
Evaluation of extramedullary leukemia, especially involvement in CNS, is necessary. CNS status at diagnosis is defined as follows: CNS1, no detectable blast cells in cerebrospinal fluid (CSF); CNS2, fewer than five leukocytes per μL with detectable blasts; and CNS3, the presence of overt CNS leukemia [75]. Although CNS2 had an adverse prognostic impact on CNS relapse in previous pediatric ALL trials, it has lost its prognostic effect in contemporary trials that include more effective systemic and CNS-directed treatment. Traumatic lumbar puncture at diagnosis is an issue, because it could cause iatrogenic CNS leukemia by mixing patients’ CSF with bloods with abundant circulating blasts, thus lead to increased risk of CNS relapse [76, 77]. In order to reduce the risk of traumatic lumbar puncture at diagnosis, correction of thrombocytopenia and coagulopathy prior to the procedure, keeping patients steady under deep sedation, and immediate administration of intrathecal therapy (e.g., methotrexate) after the collection of CSF are strongly recommended [78].
2.4.3 Treatment
Principle of ALL treatment is based on “total cell kill” theory, an attempt to eradicate every leukemic blast inside the patient’s body. This is mostly accomplished by multi-agent combination chemotherapy including CNS-directed therapy. Hematopoietic stem cell transplantation (SCT) is additionally combined in a small portion of cases with very high risk of relapse. Risk stratification, means of optimizing therapy for the patients by evaluating relapse risk with known prognostic factors, has also contributed to improve survival rate in children with ALL.
Treatment algorithm of pediatric ALL proposed in the guideline of the Japanese Society of Pediatric Hematology and Oncology is shown in Fig. 2.2. Mature B-cell ALL or Burkitt ALL, usually presented with French-American-British (FAB) L3 blasts, surface antigen expression of kappa or lambda detected by flow cytometry, and presence of cytogenetic abnormalities of t(8;14)(q24;q32), t(2;8)(p11-p12;q24), or t(8;22)(q24;q11), should be treated separately with short intensive non-Hodgkin lymphoma-oriented chemotherapy [79–81]. Ph+ ALL with t(9;22)(q34;q11.2) or BCR-ABL1 is currently treated with TKI-combined chemotherapy, thus should be treated independently [51, 52, 54]. Infants (age at diagnosis below 1 year old) with ALL are another special subgroup requiring specific care [44, 48, 82]. The rest of the children with ALL should be treated with appropriate risk-stratified therapies.
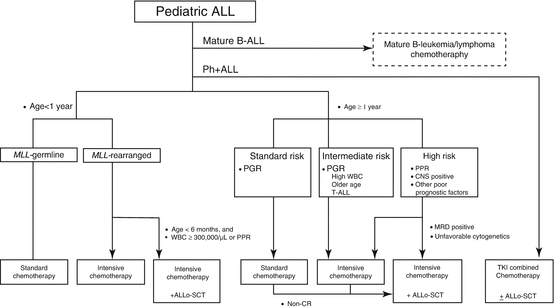
Fig. 2.2
Algorithm of treatment for children with ALL. Ph+ ALL Philadelphia chromosome-positive ALL, PPR prednisone poor responder, PGR prednisone good responder, WBC leukocyte count, CNS central nervous system, MRD minimal residual disease, TKI tyrosine kinase inhibitor, Allo-SCT allogeneic hematopoietic stem cell transplantation. [Adapted from Clinical Guideline for Pediatric Leukemia and Lymphoma (ver.3) Japanese Society of Pediatric Hematology and Oncology]
2.4.3.1 Prognostic Factors
Prognostic factors are the factors that are predictive of disease outcome and are usually used to tailor therapy (“risk-stratified therapy”) with the intention to minimize toxic events for patients with better prognosis and to maximize the treatment effect for patients with poorer prognosis. The currently well-recognized prognostic factors could be categorized into three groups.
Clinical Features at Initial Diagnosis
WBC count, which reflects tumor burden, and age at initial diagnosis have been traditionally used as most reliable prognostic factors. The NCI-Rome criteria defined patients with WBC <50,000/μL and age 1–9 years old as “standard risk” and patients with either WBC ≥50,000/μL or age ≥10 years old as “high risk” [83]. Even in the context of modern therapies, WBC count and age continue to be significant prognostic factors especially in B-ALL; however, its importance became limited in T-ALL. Infants younger than 1 year old are a special group with worse prognosis; among infants, younger age at diagnosis (e.g., age <6 months old) is an independent poor prognostic factor [44].
Related posts:
 Inherited Bone Marrow Failure Syndrome, TAM
Inherited Bone Marrow Failure Syndrome, TAM
 Childhood Aplastic Anemia
Childhood Aplastic Anemia
 Hemophagocytic Lymphohistiocytosis, Secondary
Hemophagocytic Lymphohistiocytosis, Secondary
 Primary Hemophagocytic Lymphohistiocytosis
Primary Hemophagocytic Lymphohistiocytosis
 Myelodysplastic Syndrome (MDS) and Juvenile Myelomonocytic Leukemia (JMML)
Myelodysplastic Syndrome (MDS) and Juvenile Myelomonocytic Leukemia (JMML)
 Hematopoietic Stem Cells: The Basis of Normal and Malignant Hematopoiesis
Hematopoietic Stem Cells: The Basis of Normal and Malignant Hematopoiesis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


