Fig. 2.1
Contrast-enhanced T1-weighted coronal and sagittal MR images; preoperative and 3 months postoperative
Why Does Acromegaly Fail to Be Diagnosed for Years?
Growth hormone (GH) is the most abundant of the pituitary hormones and GH secretory cells (somatotrophs) make up to 50 % of total anterior pituitary cells. GH hypersecretion is usually the result of somatotroph adenomas. Ectopic GH or GH-releasing hormone (GHrH) hypersecretion is very rare.
Clinical features of GH excess are usually progressive and insidious with a mean delay in diagnosis of 10 years or more. Mean age at diagnosis is on average 42 years, with both genders affected equally. Characteristic soft tissue proliferation with local bone overgrowth in the skull and mandible become clinically more striking over time, but often occur so gradually that they are unnoticed and unrecognized by family members and medical providers alike. Patients most commonly report growth of hands and feet and may present for orthodenture for bite abnormalities and increasing gaps between their teeth (diastema). Tufting of the distal phalanges, carpel tunnel syndrome, peripheral neuropathies, joint remodeling, and paresthesias occur. Once the epiphyses of the long bones are fused, linear growth is arrested. Spinal cord or nerve root compression from bony over growth, cardiomyopathy, and left ventricular mass enlargement leading to hypertension, cardiac arrhythmias, and valvular dysfunction are all possible. Facial features become course and the skin becomes thickened and oily with hypertrichosis and acne. Often profuse sweating associated with minimal or no physical activity is reported. Upper airway obstruction and sleep apnea commonly develop from soft tissue overgrowth. Patients report fatigue, weight gain, heat intolerance, and have often developed a decreased libido and impotence.
In the case we describe, the patient had several clear features of acromegaly that were possibly overlooked for more than 10 years. Furthermore, there was the unusual history of cosmetic procedures including a lip reduction and breast reduction surgery. A detailed history of developing symptoms such as profuse sweating, increased ring size and shoe size, and a diagnosis of obstructive sleep apnea in a tall, slim, young, and otherwise healthy patient should immediately prompt a hormonal evaluation. Unfortunately, in this case, hormonal evaluation was not undertaken when these features were developing. If a diagnosis had been made earlier, this patient would most likely have avoided the development of vision loss and joint damage and he may have avoided several cosmetic procedures. Additionally, early alterations from excess GH causes joint thickness, which can be reversed with treatment, whereas later boney complications are irreversible even with disease control and in the case of this patient may translate to permanent skeletal disabilities.
How Was the Diagnosis of GH Excess (Acromegaly) Made?
Laboratory studies collected at the time of initial evaluation showed a marked elevation in the patient’s IGF-1 levels; 1,569 ng/mL (upper limited of normal for equivalent age and sex 331 ng/mL, Table 2.1). An afternoon random GH level draw was 39.4 ng/mL (normal <5 ng/mL). Additional confirmatory testing drawn the following day (before any results available) showed that his GH level did not suppress after administration of an oral glucose load: his nadir GH level during the 2-h evaluation test was 20.9 ng/mL.
Table 2.1
Patient laboratory results collected at various treatment endpoints
Time (min) | GH (ng/mL) | IGF-1 (ng/mL; normal range) | |
|---|---|---|---|
Preoperative | Baselinea | 31 | 1,569 (53–131) |
30 | 27.5 | ||
60 | 26.5 | ||
90 | 23.8 | ||
120 | 20.9 | ||
Postoperative | Baselinea | 2.4 | 415 (53–331) |
30 | 1.2 | ||
60 | 0.8 | ||
90 | 0.9 | ||
120 | 0.9 | ||
SSA therapy | Mean 5 points GH profile | 1.6 | 385 (53–331) |
Combination therapy | _ | 131 (53–331) |
Age and gender matched IGF-1 levels are elevated in acromegaly patients and as such provides a useful screening test. A single random GH value is not a reliable screening method due to the pulsitivity of GH secretion by the pituitary. In addition to evaluating IGF-1, dynamic testing of GH is needed for confirmation of GH excess in most cases. Failure of GH to suppress after a 75 g oral glucose load to <1.0, or <0.4 ng/mL (as assessed by newer assays), confirms an acromegaly diagnosis. Nevertheless, there are acromegaly cases that present with an elevated IGF-1 and a suppressible GH after glucose administration. This test is less reliable in the setting of uncontrolled diabetes mellitus (DM), which is a frequent comorbidity in acromegaly patients. In the case we present, the patient had a significantly elevated IGF-1 level, which, in corroboration with his multiple clinical features and symptoms, secured a diagnosis of acromegaly. An oral glucose tolerance test (OGTT) substantiated these findings, but in retrospect, an OGTT was not absolutely necessary diagnostically in this particular patient.
What Additional Evaluation Should Be Performed?
After having determined acromegaly as a diagnosis or when evaluating any patient found to have a pituitary macroadenoma, a full evaluation of pituitary function is recommended. It is important to also evaluate prolactin (PRL) levels when making a new acromegaly diagnosis. This patient’s PRL was found to be mildly elevated at 29 ng/mL (normal range, 3–13 ng/mL). This was felt consistent with mass effect on the pituitary stalk from the adenoma with associated interruption of the normal tonic inhibition of PRL by dopamine.
Many GH secreting tumors also secrete PRL due to somatotroph and lactotroph cells having a common progenitor cell. It is therefore helpful to distinguish a pure somatotroph adenoma from a mammosomatotroph or a tumor with two distinct cell lines. Conversely, IGF-1 levels should also be measured in any patient presenting with evidence of a prolactinoma.
Additional biochemical assessments in the case we present included: a cortrosyn stimulation test to evaluate hypothalamic–pituitary–adrenal (HPA) axis function, which revealed a baseline cortisol of 4 mcg/dL and with stimulation up to 11 mcg/dL (stimulation to <18 mcg/dL is considered an inadequate adrenal response). His baseline ACTH level was low at 11 pg/mL thus, confirming a diagnosis of central adrenal insufficiency (AI). Thyroid function evaluation revealed a low Free T4 at 0.5 ng/dL (normal range, 0.6–1.2 ng/dL) with a corresponding normal range TSH of 2 mIU/L (normal range, 0.4–4.5 mIU/L), which was clearly inappropriate for the low free T4 and indicative of central hypothyroidism. Total serum testosterone drawn in the morning was in the low to normal range with inappropriately low to normal luteinizing hormone and follicle-stimulating hormone. This biochemical evaluation was consistent with a diagnosis of probable central hypogonadism.
How Is Acromegaly Treated?
Surgery is the first-line therapy for most patients with acromegaly. In the presence of a pituitary macroadenoma with optic chiasm displacement, a surgical approach for decompression and subsequent resection or at least tumor debulking is recommended. After initiation of replacement glucocorticoid (GC) therapy, the patient underwent semi urgent transsphenoidal surgery (TSS) due to his VF deficit. His pathology was positive for pituitary tumor cells that stained strongly (>90 %) for GH, and PRL was found in scattered cells in the sample (<10 %). Staining for other pituitary hormones was negative. The pathology report described cytokeratin staining as occurring in round perinuclear structures called fibrous bodies and as such his tumor was classified as a sparsely granulated somatotroph adenoma (Fig. 2.2).
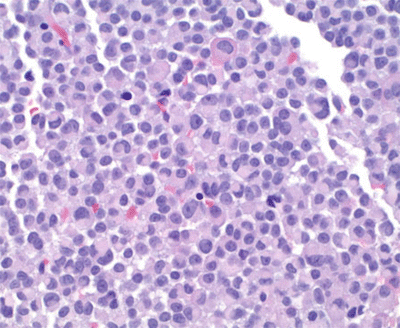
Fig. 2.2
Immunostaining for tumor cell types (magnification ×400). Sparsely granulated adenoma: cytokeratin immunostaining highlights fibrous bodies, a histological “dot–like” appearance, typically seen in these sparsely granulated tumors (cytokeratin-IHC stain)
In experienced hands, surgical remission from acromegaly can be achieved in 70–80 % of cases with microadenomas and <50 % of patients with macroadenomas at presentation. However, 40–60 % of patients surgically treated will, at some time, experience persistent or recurrent disease which requires addition of either medical or radiation therapy.
After surgery, despite a decrease in GH level within 24 h, normalization of IGF-1 level can take up to 3 months or more. Thus a diagnosis of remission vs. persistent disease should not be determined any earlier than 2–3 months postoperatively. It is apparent from several new epidemiologic studies that previous criteria for cure or remission were loosely defined. Based on new data associated with mortality rates in retrospective studies, new cut-offs have been defined: normal age and sex IGF-1 and GH suppression after a 75 g oral glucose load to <0.4 ng/mL (using newer assays).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


