Fig. 7.1
Peptide structure of Somatostatin
GH secretion is also regulated by ghrelin, a growth hormone secretagogue-receptor ligand [11] that is mainly synthesized in the gastrointestinal tract, particularly the stomach, but is also found in the central nervous system. Gastric expression of ghrelin is reduced following feeding and increased by fasting and may play a role in the regulation of GH secretion with eating [12].
IGF-I
IGF-I is responsible for most of the growth promoting actions of growth hormone (Fig. 7.2) and exerts a negative feedback on the synthesis and secretion of growth hormone [13, 14]. It is a single chain protein consisting of 70 amino acids cross-linked by three disulfide bridges, with a molecular mass of 7.6 kDa and is encoded by a gene on the long arm of chromosome 12 at position 23.2. IGF-I is secreted by many tissues, including adipose tissue, bone, muscle and kidney, and acts in an endocrine, autocrine and paracrine manner (Fig. 7.2). In mice, knockout of hepatic expression of IGF-I gene reduces circulating IGF-I levels by 80 % but has little impact on linear growth emphasizing the importance of its autocrine and paracrine actions. The A and B domains of IGF-I are homologous to the A and B chains of insulin, but the C domain shares no sequence homology [15]. IGF-I binds to a specific transmembrane tyrosine kinase receptor with activation leading to the phosphorylation of numerous substrates including the insulin receptor substrate family of proteins.
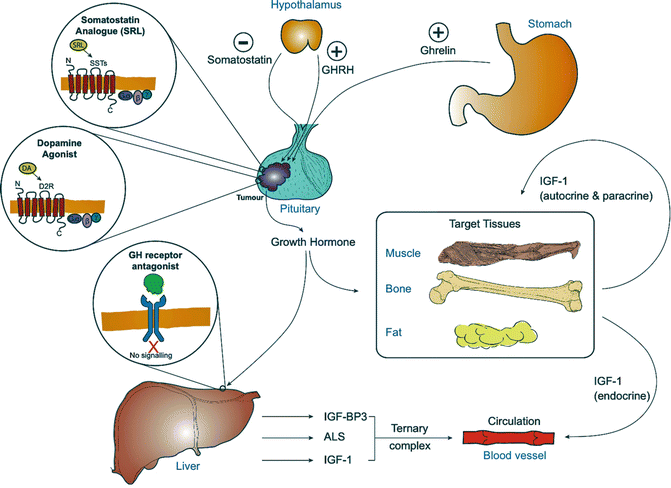
Fig. 7.2
The growth hormone-IGF-1 axis in acromegaly secondary to a pituitary tumour, indicating the site of action of the drugs used in the treatment of acromegaly
IGF-I circulates in the plasma bound to a specific family of binding proteins (IGFBPs). These binding proteins act as circulating carriers and transport the IGF-I out of the vascular compartment, deliver IGFs to specific cell types, and modulate IGF binding to receptors and growth-promoting function [16]. The majority of circulating IGF-I is in a tertiary complex formed from IGF-binding protein 3 (IGFBP3) and acid labile subunit (ALS) [17], which cannot cross the vascular epithelial layer and serves as a reservoir of IGF-I within the circulation (Fig. 7.2). Free IGF-I has a half-life of around 12 min, in contrast to over 12 h for the tertiary complex [17].
Circulating levels of IGF-I are highest in late adolescence and decline during adulthood. The levels are high in pregnancy and lower in patients with liver disease, hypothyroidism and poorly controlled diabetes [18].
Acromegaly
Clinical Features
Acromegaly is a condition defined and recognized for its phenotype which reflects the widespread expression of growth hormone receptors (Table 7.1).
CNS | Respiratory |
Headache | Sleep apnoea |
Visual field defects | Impaired respiratory function |
Cranial nerve palsies | |
Musculoskeletal | Gastrointestinal |
Prognathism | Colonic polyps |
Frontal bossing | Colonic malignancy |
Maxillary widening | Visceromegaly |
Jaw malocclusion | Endocrine and metabolic |
Increased soft tissue thickness including | Hyperprolactinemia |
Hands and feet | Menstrual abnormalities |
Arthritis and arthralgia | Galactorrhoea |
Carpal tunnel syndrome | Reduced libido |
Skin | Hypopituitarism |
Skin tags | Thyroid goitre |
Sweating | Insulin resistance |
Oily skin | Impaired glucose tolerance |
Acanthosis nigricans | Diabetes mellitus |
Cardiovascular | Hypertriglyceridemia |
Hypertension | Hypercalciuria |
Left ventricular hypertrophy | |
Cardiomyopathy | |
Congestive cardiac failure | |
Arrhythmias |
CNS
Patients may present due to mass effect from a large tumour such as headaches, visual field defects and cranial nerve palsies due to cavernous sinus invasion.
Musculoskeletal
Soft tissue growth and skeletal enlargement cause changes in the physical appearance that can be subtle and easily missed in the early stages of the disease. Changes in the face include prominence of the forehead and orbital ridges with thickening of the skin leading to frontal bossing, wide and thickened nose, prominent cheekbones, thickening of the lips, macroglossia, mandibular overgrowth with prognathism, maxillary widening, separation of the teeth and malocclusion of the jaw. It is often useful to compare old and current photographs, as the changes are very insidious.
Patients may report an increase in shoe size or ring size. Enlargement of hands results in the so-called “spade-like” appearance and the palms have a typical doughy consistency.
Accelerated degenerative changes in the weight bearing joints like the hips, knees and spine lead to a degenerative arthropathy, with arthralgia being a very common complaint. Large joint arthropathy occurs in 70 % patients secondary to the thickening of periarticular cartilaginous and fibrous tissue. 50 % patients have an axial arthropathy with restriction of movement and joint deformity [1], commonly affecting the lumbar area. Carpal tunnel syndrome occurs in 30–50 % of patients and is often bilateral but up to 80 % patients may show electrophysiological evidence of median neuropathy due to increased oedema of the median nerve in the carpal tunnel, rather than extrinsic compression from an increased volume of carpal tunnel contents [19]. Muscle hypertrophy may be present but the muscles may be weaker [20]. The thorax may be deformed due to protrusion of the lower portion of the sternum and divergence of the ribs [21].
Skin Changes
Up to 70 % of patients have oily skin with sweating being a very common symptom. The skin is thickened due to glycosaminoglycan deposition. Pigmented skin tags over the trunk are common. Acanthosis nigricans may develop in patients with severe acromegaly.
Cardiovascular System
Cardiovascular complications are a major cause of mortality in acromegaly with up to 60 % patients dying from cardiovascular disease [22], although there is no evidence of an excess of ischaemic heart disease. Hypertension affects approximately one-third of the patients with acromegaly and is associated with an expanded plasma volume, but the mechanism is ill-understood as there is no evidence that the renin–angiotensin system or catecholamines are involved in the pathogenesis [22, 23]. Kamenicky et al. demonstrated that GH with IGF-1 stimulates epithelial sodium channel mediated sodium transport in the late distal nephron, providing insight into the pathogenesis of sodium retention in acromegaly. Insulin resistance and diabetes possibly play a role in the development of hypertension [24].
Initial cardiac involvement is asymptomatic and concentric cardiac hypertrophy has been known to occur early in the condition [25, 26]. This hypertrophy is followed by a diastolic dysfunction and finally systolic dysfunction [27]. All this can lead to cardiac failure with features of a dilated cardiomyopathy. Arrhythmias are more frequently reported in patients with acromegaly, especially during exercise and these include ectopics, paroxysmal atrial fibrillation, paroxysmal supraventricular tachycardia, bundle branch blocks and ventricular tachycardia [28]. There is also an increased prevalence of valvular disorders in patients with acromegaly [29] and this risk increases with time from onset [30].
Respiratory System
Respiratory complications contribute to approximately 25 % of mortality seen with acromegaly. The respiratory problems are secondary to changes affecting facial bones and soft tissues, respiratory cartilages, lung volumes, shape of the ribcage and respiratory muscle activity [22]. The two main issues encountered are sleep apnoea and impaired respiratory function. The main cause of sleep apnoea is narrowing of the upper airways causing an obstructive sleep apnoea, although a few patients have a central cause. Impaired respiratory function is due to anatomical alterations in the chest wall as well as changes in lung elasticity. These patients may also have kyphoscoliosis, which may worsen the respiratory problems.
Endocrine and Metabolic Features
Hyperprolactinemia is seen in about 30 % of patients due to pituitary stalk compression or co-secretion of GH and prolactin by the tumour [31]. TSH co-secretion with consequent hyperthyroidism is reported. Compression of the normal pituitary tissue by the tumour results in varying degrees of hypopituitarism in approximately 40 % of patients [32]. Menstrual irregularities or erectile dysfunction, decreased libido, secondary hypothyroidism and hypoadrenalism may be seen. Goitre is common, on occasion associated with hyperthyroidism [33].
Insulin resistance and diabetes mellitus occur secondary to the anti-insulin effects of GH [34]. Hypertriglyceridemia secondary to insulin resistance is described but interestingly total cholesterol and LDL-cholesterol levels are lower in active acromegaly and rise with treatment [22, 35]. GH stimulates 1α hydroxylase, which increases the levels of 1,25-dihydroxycholecalciferol resulting in increased intestinal absorption of calcium and urinary calcium loss and potentially hypercalcaemia [36].
Controversy still exists as to the extent of cancer risk in patients with acromegaly. A direct association between acromegaly and cancer is yet to be proven [37, 38]. Gastrointestinal tumours are the most common malignancies, which may reflect that the colon is longer in patients with acromegaly but there is also evidence that [39] recurrent colonic adenomas (but not hyperplastic polyps) showed a correlation with IGF-I levels [40]. In a retrospective cohort study, colon cancer mortality but not incidence was found to be higher [41]. It is recommended that patients who have acromegaly should be offered a screening colonoscopy at baseline [42]. The frequency of repeat colonoscopy should depend on the findings of the original screening and should be according to the international guidelines for colon cancer [42].
Biochemical Confirmation of Acromegaly
The rarity of the disease means that the greatest challenge in the diagnosis of acromegaly is for the disease to enter the differential diagnosis of the presenting manifestations, and this is reflected in typically a decade lapsing between development of symptoms and confirmation of the diagnosis. Once considered, the diagnosis is usually rapidly confirmed by biochemical testing and imaging.
Biochemical Tests
An undetectable (<0.3 mcg/l) random GH measurement is good evidence against a diagnosis of acromegaly, but an elevated value is of limited value as it may be a pulse in a healthy individual. For that reason a combination of an oral glucose tolerance test and serum IGF-I measurement are required in suspected acromegaly, with results being unequivocally abnormal in patients with acromegaly.
The gold standard test for a biochemical diagnosis of acromegaly is the 75-g oral glucose tolerance test. Growth hormone level is measured at baseline and at every 30-min intervals for 2 h. In normal individuals, GH levels fall with at least one value being <0.3 mcg/L, with failure of suppression or a paradoxical rise in GH being indicative of acromegaly.
When interpreting GH data, and in particular when applying international consensus criteria to local practice, it must be appreciated that although the performance of GH assays, by some criteria, have improved, that by others their clinical applicability has deteriorated, for example the bias between different commercial assays has increased. In other words, the reported value for a given sample can vary greatly dependent on the assay methodology and it is erroneous to ascribe undue biological significance to a given consensus cut-off such as 0.3 mcg/L, but rather recognize that it is “best-guess” that is assay bias dependent. The origins of assay bias are discussed elsewhere [43].
GH can fail to suppress during an OGTT in patients with uncontrolled diabetes mellitus, liver or renal disease, patients receiving oestrogen, during pregnancy and late adolescence, malnutrition and anorexia, but in combination with IGF-I measurement and examination of the patients there is rarely a problem [18]. It should not be difficult to distinguish anorexia from acromegaly!
The utility of IGF-I measurement is hindered by concerns over the quality of some commercial assays and their reference ranges. This is a rare problem with the diagnosis of acromegaly as the levels are normally unequivocally elevated—an IGF-I within the reference range in newly diagnosed acromegaly is very rare. A greater problem is apparently elevated IGF-I levels in patients who do not have acromegaly. The explanation of such findings is uncertain and probably lies with limitations of assay design and reference ranges and often results in extensive investigation to disprove the diagnosis. All IGF-I assays must first be thoroughly validated and standardized to the new International IGF-I standard IS02/254. Furthermore, many of the clinical frustrations related to IGF-I assessment are a consequence of the inadequacies of the reference range. Brabant et al. established the gold standard reference range by collecting data for 3,961 healthy subjects [44].
IGF-I levels may vary, typically being low in liver and renal dysfunction and uncontrolled diabetes mellitus. Nutrition, circadian rhythm, oestrogen, insulin, glucocorticoid therapy and thyroxine levels can affect IGF-I levels.
Imaging
Magnetic resonance imaging (MRI) with gadolinium contrast is the imaging modality of choice and can detect tumours as small as 2 mm; although more than 75 % of patients have a pituitary tumour >10 mm in diameter. When an extra-pituitary source of GH or GHRH is suspected, abdominal and chest CT with or without MRI may be needed. In the rare cases of ectopic GHRH the pituitary typically has a hyperplastic appearance.
Functional Pituitary Testing
As with any pituitary tumour, evidence of hypopituitarism must be sought using standard protocols. Elevated prolactin levels could be secondary to compression of the pituitary stalk or due to the tumour co-secreting GH and prolactin. Prolactin levels >5,000 mU/L indicates co-secretion; with more minor degrees of hyperprolactinaemia the aetiology is less certain. Elevated GH levels suppress hepatic cortisol binding globulin (CBG) expression and lower the circulating CBG levels. Therefore in active acromegaly measurement of total serum cortisol may underestimate free cortisol and after successful surgery apparent recovery in circulating cortisol can be seen, which is actually a consequence of an increase in CBG.
Visual field testing should be undertaken in all patients with macroadenomas.
Morbidity, Mortality and Defining Disease Control
The excess mortality of acromegaly [45] is multifactorial in origin with important contributors being cardiomyopathy, hypertension, hyperglycaemia or diabetes and sleep apnoea.
There is accumulating evidence that vigorous treatment and good biochemical control (see below) not only reduces morbidity but also restores life expectancy to normal [45–50].
GH values and IGF-I should be used as complementary tests to assess for disease control and activity. It is not uncommon to get a discrepancy between the GH and IGF-I levels [51], most commonly with normal GH levels and an elevated IGF-I although the reverse can be seen. Factors that can lead to discrepancies include prior radiotherapy, and gender. Women have lower IGF-I levels for a given GH level than men, an effect exaggerated by the use of oral oestrogen [52]. GH levels fall rapidly after surgery but IGF-I can take in excess of 3 months to reach its nadir.
Remission Criteria
The definition of disease control has become more rigorous over the past decade. This is partially due to the recognition that mortality associated with acromegaly reduces with an improvement in the biochemistry.
Optimal disease control is currently defined as a random GH level of <1 μg/L using an ultrasensitive assay or nadir GH of <0.4 μg/L on OGTT; and an IGF-I in the age-adjusted normal range [52] (Table 7.2). When there is discrepancy between GH and IGF-I values, multiple GH sampling (three to five times over 2 h) is suggested. For patients on medical treatment with a somatostatin receptor analogue or dopamine agonist, IGF-I and random GH measurements may suffice [52]. An OGTT is not helpful for monitoring response in patients receiving medical treatment. Patients on treatment with a growth hormone receptor antagonist are monitored using IGF-I levels only.
Outcome | Criteria | Management |
|---|---|---|
Active acromegaly | Random GH >1 μg/L and nadir GH after OGTT ≥0.4 μg/L | Frequent MRI |
Elevated IGF-1 | Monitor and actively treat comorbidities | |
Clinical features of active disease | Actively treat or consider change of treatment | |
Controlled acromegaly | Random GH <1 μg/L or nadir GH after OGTT <0.4 μg/L | Periodic but less frequent MRI |
Age–sex normalized IGF-1 | No change to treatment, consider reducing SSA dose |
Management
Advances in pituitary surgery, the development of new pharmaceutical interventions and refinements in radiotherapy mean that tumour control and biochemical remission can be achieved in the overwhelming majority of patients. The challenge is optimizing the treatment algorithm recognizing local variations in access to the treatment modalities and ensuring minimal treatment related morbidity.
Surgery
The transsphenoidal microsurgical approach is the appropriate initial treatment in the great majority of patients. In expert hands, approximately 80 % and 50 % of patients with micro- and macroadenomas, respectively, achieve normal IGF-I levels [53, 54]. While the merits of endoscopic approach with or without intra-operative MRI are debated, unquestionably the choice of the surgeon is crucial, illustrated by the variation in published outcomes between centres in the UK [55]. Cure rates as low as 17.8 % have been reported in one centre in which multiple neurosurgeons were undertaking a small number of cases each [56], which improved to 67 % with a restructuring of the service to one dedicated pituitary surgeon [57]. There is also a plethora of evidence of morbidity and mortality being lower in the hands of specialist pituitary surgeons [58].
Complications of pituitary surgery include diabetes insipidus, which is usually transient. CSF rhinorrhoea and rarely meningitis may occur. Alteration in the sense of smell, epistaxis and sinusitis may be seen. The syndrome of inappropriate ADH secretion (SIADH) with hyponatremia may be seen transiently in the post-operative period. It is typically seen 7 days following surgery and usually resolves with fluid restriction. The main long-term complication is hypopituitarism, which may be seen in 10–20 % cases [59] depending on tumour size and the surgeon’s experience. Damage to the optic nerve or to the carotid artery should be seen very rarely (<1 %) and mortality should be less than 0.5 %.
Radiotherapy
Pituitary radiotherapy is very effective at controlling tumour growth and, with time, controls GH secretion and normalizes IGF-I. Conventional multi-fractionated, 3-field radiotherapy delivering 4,500 cGy has fallen out of favour, particularly in younger people, because of the risk of cerebrovascular accidents but still has a place in the treatment of patients with large and growing residual tumour after surgery. GH values can be anticipated to reduce by up to 50 % in the first 2 years, followed thereafter by a continuing slow decline [60]. IGF-I normalization is also seen in 60 % patients after 10 years, so somatostatin analogue therapy is often required for many years post-irradiation. Hypopituitarism is the most common complication. In one large series, 10 years after irradiation, 27 % developed TSH deficiency, 18 % developed FSH/LH deficiency and 15 % developed corticotropin deficiency [60].
Stereotactic high dose pituitary irradiation has in many centres supplanted conventional radiotherapy most frequently being delivered by the Gamma Knife®. These techniques require precise delineation of the tumour to ensure there is minimal surrounding tissue exposure and therefore are more suitable for smaller volumes residual tumour beyond the reach of the surgeon.
Among case series of GH adenomas treated with radiosurgery, tumour control was attained in 97 % cases with hormonal remission rates varying from 17 to 96 % [59]. Hypopituitarism is the most common side effect.
Medical Treatment
Dopamine Receptor Agonists
Dopamine receptor agonists, acting through the D2 receptor (Fig. 7.2), were the first effective medical therapy for acromegaly and continue to have a place in its treatment, often in combination with somatostatin analogues, particularly as they have the twin virtues of being relatively inexpensive and orally administered. Early studies with bromocriptine documented suppression of GH levels to <5 μg/L in about 15 % patients when used in high doses of up to 20 mg/day with corresponding reductions in symptoms like soft tissue swelling and headache [61]. Cabergoline is a long-acting ergot-derived dopamine agonist that has superseded bromocriptine as it is more potent and better tolerated. Doses of up to 1 mg/day normalize IGF-I in up to 30 % patients [62]. Cabergoline is probably most effective in patients with prolactin co-secretion when it is likely to also induce some tumour shrinkage [63]. Cabergoline is not licensed for the treatment of acromegaly and dose finding and large scale prospective studies have not been undertaken so it is probable that the potential of cabergoline has not been fully exploited either in terms of biochemical control or tumour shrinkage. Side-effects include gastrointestinal discomfort, nausea, vomiting, dizziness, headache and postural hypotension but in most patients these are manageable by slow dose titration. The more concerning side effects include induction of depression or mania often in patients with a prior history or very rarely development of gambling, alcohol and sex addiction in patients with no history of previous psychiatric problems. There is evidence in patients with Parkinson’s disease, of ergot-derived dopamine agonists causing cardiac valve fibrosis with the risk being related to cumulative dose [64] and although the doses used in endocrine patients are much smaller and there is a dearth of evidence of a problem in patients with pituitary adenomas, annual echocardiograms are recommended.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


