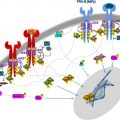
The finding of somatically acquired uniparental disomy, where both copies of a chromosome pair or parts of chromosomes have originated from one parent, has led to the discovery of several novel mutated genes in myeloproliferative neoplasms and related disorders. This article examines how the development of single nucleotide polymorphism array technology has facilitated the identification of regions of acquired uniparental disomy and has led to a much greater understanding of the molecular pathology of these heterogeneous diseases.
- •
Uniparental disomy is where both copies of a chromosome pair or parts of chromosomes have originated from one parent.
- •
Acquired uniparental disomy in cancer is a mechanism by which adventitious mutations are amplified leading to a growth advantage of these cells.
- •
Acquired uniparental disomy is now understood to be common in leukemia and renders a malignant or premalignant cell homozygous for a pre-existing mutation.
- •
Myeloproliferative neoplasms are clonal hematopoietic stem cell disorders characterized by overproliferation of one or more myeloid cell lineages in the bone marrow and increased numbers of mature and immature myeloid cells in the peripheral blood.
- •
Single nucleotide polymorphism arrays use the most frequent type of variation in the human genome and have enabled the rapid identification of uniparental disomy.
- •
Identification of tracts of recurrent acquired uniparental disomy, especially in hematologic malignancies, has led to identification of novel driver genes and therefore highlighted new pathways for targeted therapy.
Introduction
Cancer genomes are characterized by instability and a progressive accumulation of genetic aberrations. Loss of heterozygosity (LOH) is one such aberration and is widely recognized as a hallmark of cancer genomes. LOH is most commonly caused by whole and partial chromosomal loss as a consequence of aneuploidy or somatically acquired deletions but in recent years it has become apparent that LOH may also be caused by uniparental disomy (UPD).
UPD refers to the situation in which both copies of a chromosome pair or parts of chromosomes have originated from one parent. When it occurs, UPD is often constitutional and arises from errors at meiosis I or meiosis II, the latter giving rise to isodisomy whereby the affected region is genetically identical. Constitutional isodisomy is frequently associated with developmental disorders caused by the abnormal expression of imprinted genes in the affected regions. In cancer, UPD is acquired somatically and was first associated with the development of retinoblastoma. It is now known that acquired UPD (aUPD; also known as acquired isodisomy or copy number neutral LOH) in cancer is a mechanism by which adventitious mutations are amplified, leading to a growth advantage of these cells. Acquired UPD is common in solid cancers and leukemia and identification of tracts of recurrent aUPD, especially in hematologic malignancies, has led to identification of novel driver genes.
This article describes how single nucleotide polymorphism (SNP) array technology has greatly facilitated the identification of regions of aUPD and led to the identification of novel mutations in myeloproliferative neoplasms (MPNs) and related disorders.
UPD and the importance of SNP array technology
Determining whether or not UPD is present is not possible using conventional cytogenetics, fluorescent in situ hybridization, or comparative genomic hybridization because there is no change in copy number and these techniques are usually unable to distinguish between maternal and paternal chromosomes. Before the completion of the Human Genome project and the wealth of SNP data that derived from it, the identification of UPD was cumbersome, involving restriction fragment length polymorphism analysis where only small regions of the genome could be interrogated or microsatellite analysis, which laboriously provided low resolution over the genome. The advent of SNP array technology meant that genetic variation over the entire genome could be identified rapidly at much higher resolution than was previously possible.
SNP arrays work by hybridizing the fragmented and fluorescently labeled sample DNA to immobilized oligonucleotide probes on glass plates or in solution. The probes are regularly spaced over the entire genome and used to identify the genotypes at specific polymorphic loci. Laser capture then identifies the ratios of fluorescent sample annealed to the probes.
The first SNP array experiments looking at cancer genomes had only 600 to 1000 probes, and involved polymerase chain reactions of each SNP locus. Now preparation of the sample can be done in one tube with the number of probes exceeding 1 million. This has allowed for even higher throughput and resolution of the whole genome. SNP array technology can be used routinely to screen many patients for recurrent regions of aUPD and this information can be used in two ways: to examine if these regions are associated with prognosis, and to determine the minimal affected regions (MARs) of aUPD and thereby target genes that might be mutated.
UPD and the importance of SNP array technology
Determining whether or not UPD is present is not possible using conventional cytogenetics, fluorescent in situ hybridization, or comparative genomic hybridization because there is no change in copy number and these techniques are usually unable to distinguish between maternal and paternal chromosomes. Before the completion of the Human Genome project and the wealth of SNP data that derived from it, the identification of UPD was cumbersome, involving restriction fragment length polymorphism analysis where only small regions of the genome could be interrogated or microsatellite analysis, which laboriously provided low resolution over the genome. The advent of SNP array technology meant that genetic variation over the entire genome could be identified rapidly at much higher resolution than was previously possible.
SNP arrays work by hybridizing the fragmented and fluorescently labeled sample DNA to immobilized oligonucleotide probes on glass plates or in solution. The probes are regularly spaced over the entire genome and used to identify the genotypes at specific polymorphic loci. Laser capture then identifies the ratios of fluorescent sample annealed to the probes.
The first SNP array experiments looking at cancer genomes had only 600 to 1000 probes, and involved polymerase chain reactions of each SNP locus. Now preparation of the sample can be done in one tube with the number of probes exceeding 1 million. This has allowed for even higher throughput and resolution of the whole genome. SNP array technology can be used routinely to screen many patients for recurrent regions of aUPD and this information can be used in two ways: to examine if these regions are associated with prognosis, and to determine the minimal affected regions (MARs) of aUPD and thereby target genes that might be mutated.
Advantages and disadvantages of aUPD analysis by SNP array technology
For any given technique there are advantages and disadvantages, and aUPD analysis in leukemia with SNP arrays is no exception. SNP arrays, unlike metaphase cytogenetics, are not reliant on cell growth to yield detailed data on karyotype and although its throughput, resolution, and detailed mapping are vastly superior to microsatellite analysis and metaphase cytogenetics, SNP arrays cannot distinguish between one clone with several defects from several distinct clones. The technique effectively detects copy number changes (deletions or amplifications) but cannot detect balanced translocations that may, for example, be strong indicators for specific targeted therapies. Thus, the commonly used term “SNP array karyotyping” is something of a misnomer.
Because SNP arrays were originally designed to look at population variation and germ line mutations, there were several problems that needed to be overcome before successful analysis of cancer samples could be accomplished. The malignant clone size in clinical samples can be small and may be masked by a variably sized background of contaminating normal cells. This can make aUPD difficult to ascertain using conventional analysis software and therefore several algorithms were designed to enable identification of low levels of aUPD. The resolution of these techniques depends in part on the size of the affected region, but it should be possible to detect larger regions in samples with only 20% to 30% tumor cells, although a higher purity is preferred.
To unambiguously detect somatically acquired UPD it is critical to compare tumor (or tumor enriched) DNA with constitutional DNA (eg, derived from T-cells, buccal epithelia, fibroblasts, or remission samples). In the absence of constitutional DNA the possibility of an inherited region of homozygosity cannot be excluded. Indeed, analysis of lymphoblastoid cell lines derived from healthy individuals identified contiguous homozygous tracts greater than 5 Mb in nearly 10% of cases with some cases showing multiple homozygous tracts across the genome, probably as a result of consanguinity. These regions were confirmed in peripheral blood samples in the subset of cases that were analyzed. In healthy individuals, runs of homozygosity greater than 20 Mb are very uncommon and therefore we have used this as a cutoff for defining likely regions of aUPD in our analysis. Other studies have used much smaller cutoffs and in the absence of constitutional DNA it is likely that many of these regions are not derived by aUPD at all, but are simply identical by descent and of no pathogenetic significance. It is probably more appropriate to refer to these regions as copy number neutral runs of homozygosity if material is not available to prove somatic acquisition.
Identification of novel mutations in MPN and MDS/MPN using regions of aUPD
Somatically acquired UPD is now understood to be common in leukemia and renders a malignant or premalignant cell homozygous for a pre-existing mutation. Reduction to homozygosity as a consequence of aUPD was initially thought to be only a mechanism for inactivation of tumor suppressor genes ; however, identification of aUPD in leukemia showed that oncogenic mutations are also targeted.
MPNs are clonal hematopoietic stem cell disorders characterized by overproliferation of one or more myeloid cell lineages in the bone marrow and increased numbers of mature and immature myeloid cells in the peripheral blood. Excess proliferation is frequently associated with splenomegaly and cardiovascular complications and increased risk of transformation to acute leukemia. MPNs are categorized into subtypes based on specific morphologic, hematologic, and laboratory parameters, the best characterized being the four so-called classic MPNs: (1) polycythemia vera (PV), (2) essential thrombocythemia (ET), (3) primary myelofibrosis (PMF), and (4) chronic myeloid leukemia. In addition, some MPN cases have overlapping features with myelodysplastic syndromes (MDSs) and are classified separately as MDS/MPN, such as atypical BCR-ABL negative chronic myeloid leukemia and chronic myelomonocytic leukemia.
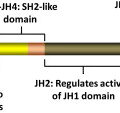
PV was the first hematologic malignancy to be associated with aUPD with the finding of a recurrently affected region at chromosome 9p. However, its significance would remain obscure until this region was associated with the oncogenic V617F mutation in the JAK2 gene in PV. This acquired, single point mutation in JAK2 was described in 95% of patients with PV, and 50% of patients with ET and PMF. Acquired UPD at 9p results in a population of cells that are homozygous for V617F, and this is seen most commonly in PV and PMF but is rare in ET.
SNP array analysis of MDS/MPN revealed that these were a heterogeneous group of diseases, accompanied by higher genetic instability than was previously thought. Moreover, regions of recurrent aUPD in otherwise karyotypically normal patients were a common finding, suggesting the presence of novel mutated genes in these patients. Acquired UPD at 11q was one of the most common findings in these diseases, and candidate gene screening of the MAR revealed that the target in most of these cases was the Casitas B-lineage lymphoma ( CBL ) gene. CBL is a negative regulator of tyrosine kinase signaling. In its positive role CBL binds to activated receptor tyrosine kinases by its N-terminal tyrosine kinase binding domain and serves as an adaptor by recruiting downstream signal transduction components, such as SHP2 and PI3K. However, the RING domain of CBL has E3 ligase activity and ubiquitinylates activated receptor tyrosine kinases on lysine residues, a signal that triggers internalization of the receptor/ligand complex and subsequent recycling or degradation. CBL mutations had already been found in occasional cases of acute myeloid leukemia but they were much more common in MDS/MPN at a frequency of about 10% and were a new class of mutation in these diseases.
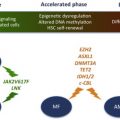
Despite SNP array refining candidate regions of aUPD to relatively small regions of the genome, some MARs contained hundreds of genes. However, SNP array data contain information on UPD and deletions. Combining both of these data sets allowed identification of additional targets. The first example of this approach focused on 4q aUPD in MDS/MPN, where focal microdeletions led to the identification of the TET2 gene on 4q24 and further analysis showed this to be commonly mutated in many different subtypes of MDS and MPN. Although its function at the time was unknown, TET2 is involved in epigenetic regulation; specifically, TET2 mediates the hydroxylation of 5-methylcytosine to 5-hydroxymethylcytosine in DNA. TET2 mutations are inactivating and often seem to be early events in the development of MPN and MDS/MPN. Their prognostic value, if any, remains a matter of debate and may depend on the precise disease subtype.
In contrast, acquired UPD of 7q, found in 10% of MDS/MPN, was linked with a poor prognosis before mutation identification. Enhancer of zeste 2 ( EZH2 ) was subsequently identified as the target, again by the finding of focal microdeletions involving this gene. EZH2 interacts with EED, SUZ12, and RBBP4 (also known as RbAp48) to form the polycomb repressive complex (PRC)-2, which functions to initiate epigenetic silencing of genes involved in cell fate decisions. EZH2 is the catalytic component of PRC2 and specifically methylates lysine 27 of histone H3. Trimethylated H3 (H3K27me3) serves as a signal for recruitment of further proteins, including PRC1, which maintains a silenced state. Although EZH2 had been implicated in a variety of cancers, inactivation of the gene through mutation had not before been observed. Mutations in EZH2 , like the aUPD of 7q that amplifies it, is a poor prognostic marker in MPN and MDS/MPN. Identification of EZH2 mutations in these diseases has led to other members of PRC2 to be identified as mutated in MPN and other leukemias, underscoring the importance of genes involved in epigenetics in the genesis of leukemia. Indeed, next-generation sequencing has taken this work forward and identified other mutated epigenetic genes in MDS/MPN, such as UTX and DNMT3A. Several other regions of aUPD have been identified in MDS/MPN and it is likely that the putative targets of these events will be identified soon by next-generation sequencing approaches.
In addition to reducing mutations to homozygosity, it is possible that aUPD is associated with changes in expression levels of the genes in the affected region because some genes have expression dependent on the parent of origin, whereas others are monoallelically expressed. Acquired UPD may therefore have other as yet uncharacterized consequences that might, for example, give rise to interindividual differences within patient subgroups that could be dependent on individual genetic variation and the position of the mitotic recombination breakpoint that led to aUPD.
Mechanisms underlying aUPD
There are two mechanisms by which aUPD is thought to occur, broadly based on the type of aUPD. Segmental aUPD is thought to occur because of a reciprocal exchange of chromosomal material during mitosis, known as “mitotic recombination,” which is often evident by homozygosity from the point of crossing over to the telomere ( Fig. 1 ) but may give rise to interstitial aUPD if there are two points of recombination. Whole chromosome aUPD is likely to arise from nondisjunction, where an attempt is made to correct for loss of the chromosome material using the remaining chromosome copy as a template. The net result, however, is that the two chromosomes harboring the mutation segregate into the same daughter cell and provide it with a growth advantage.