Platelets play a critical role in hemostasis. When the vascular endothelium is disrupted, platelets adhere to the subendothelium and initiate primary hemostasis. The details of normal platelet physiology and function are presented in Chapter 26 . Excessive bleeding occurs if primary hemostasis is abnormal because platelets are either deficient in number or defective in function. Acquired platelet defects, both quantitative and qualitative, are discussed in this chapter, and the corresponding inherited disorders are discussed in Chapter 30 .
The normal circulating platelet count for all ages ranges from 150,000 to 400,000/µL. Circulating platelets constitute two thirds of total body platelets; the remaining platelets are located within the spleen. Platelets exhibit marked heterogeneity in size. Two factors have been proposed to account for this heterogeneity. First, as platelets age, they may become smaller as a result of fragmentation or loss of granule contents or membrane proteins. Second, megakaryocytes produce platelets of varying size. In thrombolytic states megakaryocytes preferentially produce large platelets, a phenomenon analogous to stress erythropoiesis. The average life span of platelets is 7 to 10 days, although survival of transfused platelets in a thrombocytopenic recipient is reduced proportionately to the severity of the thrombocytopenia. These findings suggest either that there is increased platelet utilization in thrombocytopenic states or that a fixed number of platelets are removed from the circulation each day, irrespective of the platelet count.
Quantitative Platelet Abnormalities
Thrombocytopenia
The clinical manifestations of thrombocytopenia typically involve the skin or mucous membranes and include petechiae, ecchymoses, prolonged bleeding at incision or venipuncture sites, epistaxis, gastrointestinal hemorrhage, hematuria, and menorrhagia. Intracranial hemorrhage (ICH) may occur but is rare. As discussed in Chapter 29 , the deep muscle hematomas and hemarthroses typically seen in individuals with deficiencies of factor VIII or factor IX generally do not occur with platelet disorders. Causes of thrombocytopenia fall into three broad categories: platelet sequestration (usually in the spleen), increased platelet destruction, and decreased platelet production. The differential diagnosis of thrombocytopenia in children is outlined in Box 34-1 . Discriminating among these diagnoses is important because the cause of thrombocytopenia affects the choice of therapy.
Destructive Thrombocytopenias
Primary Platelet Consumption Syndromes
Immune thrombocytopenias
Acute and chronic ITP
Autoimmune diseases with chronic ITP as a manifestation
Cyclic thrombocytopenia
ALPS and its variants
SLE
Evans syndrome
Antiphospholipid antibody syndrome
Neoplasia-associated immune thrombocytopenia
Thrombocytopenia associated with HIV
Neonatal immune thrombocytopenia
Alloimmune
Autoimmune (e.g., maternal ITP)
Drug-induced (including HIT)
Posttransfusion purpura
Allergy and anaphylaxis
Nonimmune thrombocytopenias
Thrombocytopenia of infection
Bacteremia or fungemia
Viral infection
Protozoan infection
Thrombotic microangiopathic disorders
TTP
HUS
BMT–associated microangiopathy
Drug-induced
Platelets in contact with foreign material
Congenital heart disease
Drug-induced via direct platelet effects (ristocetin, protamine)
Type 2b VWD or platelet-type VWD
Combined Platelet and Fibrinogen Consumption Syndromes
DIC
Kasabach-Merritt syndrome
HLH
Impaired Platelet Production
Hereditary disorders (see Chapter 30 )
Acquired disorders
Aplastic anemia
MDS
Marrow infiltrative process
Nutritional deficiency states (Fe, folate, vitamin B 12 , anorexia nervosa)
Drug- or radiation-induced thrombocytopenia
Neonatal hypoxia or placental insufficiency
Sequestration
Hypersplenism
Hypothermia
Burns
ALPS, Autoimmune lymphoproliferative syndrome; BMT, bone marrow transplant; DIC, disseminated intravascular coagulation; HLH, hemophagocytic lymphohistiocytosis; HIT, heparin-induced thrombocytopenia; HIV, human immunodeficiency virus; HUS, hemolytic-uremic syndrome; ITP, immune thrombocytopenic purpura; MDS, myelodysplastic syndrome; SLE, systemic lupus erythematosus; TTP, thrombotic thrombocytopenic purpura; VWD, von Willebrand disease.
When a patient with thrombocytopenia is assessed, the risk of bleeding episodes should be estimated. If the risk is significant, treatment is warranted. Unfortunately, there is a lack of direct correlation between the platelet count and the risk of bleeding episodes, which confounds treatment decisions. The risk of hemorrhage is affected by many factors, such as coexisting coagulation defects, trauma, and surgery. In older children and adults, serious spontaneous bleeding does not occur until the platelet count is less than 20,000/µL. Many physicians use a platelet count of 10,000 to 20,000/µL as the threshold for intervention. This threshold was derived from a study of children with leukemia and may not be relevant for all cases of thrombocytopenia. For example, because of the increased risk for ICH in neonates, a threshold of 20,000 to 50,000/µL is often used. In addition, patients with a defect in production are more likely to have serious bleeding than those with a destructive platelet problem because in the latter patients, platelets tend to be larger and more functional.
Thrombocytopenia Reflecting Laboratory Artifact or Sequestration
Spurious Thrombocytopenia.
Thrombocytopenia may be an incidental finding. If the history and physical examination do not suggest a defect in primary hemostasis, a low platelet count may represent a laboratory artifact. Potential causes of a falsely low platelet count include platelet activation during blood collection, undercounting of megathrombocytes, or pseudothrombocytopenia as a result of in vitro agglutination by ethylenediaminetetraacetic acid (EDTA)-dependent antibodies. In a patient with artifactual thrombocytopenia, large clumps of agglutinated platelets may be found at the periphery of the blood film, or platelets may be adherent to leukocytes and form platelet “satellites” ( Fig. 34-1 ).
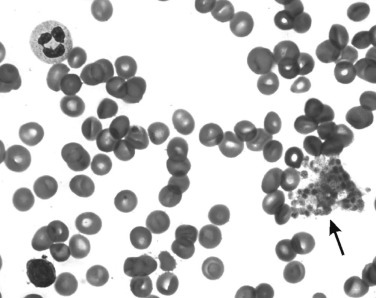
Pseudothrombocytopenia secondary to EDTA-dependent antibodies can be confirmed by repeating the platelet count with another anticoagulant (e.g., citrate, oxalate, heparin) or by preparing a blood film directly from a finger- or heel-puncture sample. The cause of EDTA-associated pseudothrombocytopenia is an immunoglobulin G (IgG) or immunoglobulin M (IgM) directed against a cryptic platelet antigen exposed only in the presence of this anticoagulant. The phenomenon is not associated with any particular pathology and may be observed in both healthy subjects and patients with diseases. In some individuals the antibodies responsible persist indefinitely, whereas in others the antibodies are transient. No abnormalities in hemostasis or thrombosis have been reported in any of these patients.
Other causes of pseudothrombocytopenia include drugs and EDTA-independent cold agglutinins. The overall incidence of pseudothrombocytopenia in hospitalized adult patients is approximately 1%, but it is less common in pediatric patients. No further evaluation or treatment is indicated for a patient documented as having a spuriously low platelet count.
Apparent Thrombocytopenia Caused by Hypersplenism.
The spleen normally retains about a third of the body’s platelets in an exchangeable pool. The fraction of platelets sequestered in the spleen increases in proportion to spleen size. Thus an apparent thrombocytopenia can result from increased pooling in an enlarged spleen—a condition referred to as hypersplenism . In patients with hypersplenism, recovery of transfused autologous platelets is only 10% to 30%, whereas in normal individuals it is 60% to 80% and in asplenic patients it is 90% to 100%. Splenic blood flow is the major determinant of the size of the exchangeable splenic platelet pool in splenomegalic states. Administration of intravenous epinephrine causes constriction of the splenic artery and results in passive emptying of platelets into the circulation. This increase in platelet count is proportionately greater in patients with splenomegaly than in normal subjects.
In general, the apparent thrombocytopenia that results from pooling in an enlarged spleen is mild (50,000 to 150,000/µL). Platelet counts of less than 50,000/µL should not be attributed to splenomegaly alone without further investigation. The degree of hypersplenism is proportional to spleen weight, whether the splenomegaly is due to congestion (e.g., cirrhosis with portal hypertension), hemolytic anemia (e.g., hemoglobin SC disease), or other causes. Because thrombocytopenia secondary to splenic pooling is not usually clinically important, no treatment is warranted, although splenectomy may be indicated for patients with severe thrombocytopenia, as can occur in Gaucher disease or other storage diseases.
Apparent Thrombocytopenia Caused by Hypothermia.
Platelets are transiently sequestered in the spleen, liver, and other organs of experimental animals subjected to hypothermia. On rewarming of the animal, these platelets return to the circulation. A similar phenomenon has been observed in hypothermic patients. Transient thrombocytopenia (platelet counts of 7000 to 62,000/µL) has been reported in hypothermic patients of various ages. Less significant thrombocytopenia has been observed in patients undergoing cardiac surgery with hypothermic perfusion. Treatment of the thrombocytopenia associated with hypothermia consists of rewarming and documentation of return of the platelet count to the normal range, which usually occurs in 4 to 10 days.
Thrombocytopenia Caused by Increased Platelet Destruction
A major etiologic classification of acquired thrombocytopenia of childhood is increased platelet destruction. Clinical conditions associated with increased platelet destruction are listed in Box 34-1 . These conditions can be further subgrouped into immune and nonimmune causes.
Immune Thrombocytopenias.
Autoantibodies, alloantibodies, or drug-dependent antibodies may associate with platelet membranes and target the cells for accelerated destruction by phagocytes of the reticuloendothelial system. An antibody mediating immune destruction of platelets may be directed against a platelet membrane antigen, or it may be part of an immune complex that binds Fc receptors on platelets.
Platelet antigens fall into two general classes. Glycoproteins that occur predominantly on platelets, such as the glycoprotein IIb/IIIa (GPIIb/IIIa) or GPIb/IX/V complexes, are often termed platelet-specific antigens . The glycoproteins (e.g., human leukocyte antigen [HLA] class I) and glycolipids (e.g., blood group ABH antigens) expressed on platelets, leukocytes, and other cell types are termed platelet-nonspecific antigens . Antibodies against platelet-specific and platelet-nonspecific antigens are responsible for a number of clinical syndromes, including autoimmune thrombocytopenia, neonatal alloimmune thrombocytopenia (NAIT), posttransfusion purpura (PTP), and platelet transfusion refractoriness. Antibodies directed against platelet integrins are common in immune thrombocytopenias. For example, autoantibodies directed against GPIIb/IIIa are seen in immune thrombocytopenic purpura (ITP) and in drug-induced thrombocytopenia. Several clinically significant alloantigens are located on GPIIb/IIIa, including HPA-1a, the human platelet antigen most frequently implicated in NAIT (discussed later). Autoantibodies against GPIb or GPIX also have been reported in patients with ITP and drug-induced thrombocytopenia.
HLA class I antigens are expressed on a wide range of cells, including platelets, and are important for the recognition of self by cytotoxic T cells. HLA-A and HLA-B antigens are strongly expressed on platelets. Alloantibodies against HLA-A or HLA-B antigens frequently form in multiparous women and multiply transfused patients, and these alloantibodies contribute to platelet transfusion refractoriness. HLA-C antigens are weakly expressed on platelets and are far less likely to induce alloantibodies, thus making it less necessary to match the HLA-C locus for donor-recipient compatibility in platelet transfusions. HLA class II antigens, which function in antigen presentation by macrophages and B lymphocytes, are not expressed on platelets.
ABH blood group antigens are carried on a number of platelet glycoproteins, including the GPIIb/IIIa and GPIb/IX/V complexes. ABH antigens are also expressed on glycolipids. In rare instances these antigens have been implicated as the cause of immune-mediated platelet destruction, but in general these platelet-nonspecific antigens play only a minor role in immune thrombocytopenias.
Platelet Antibody Testing.
A large number of assays have been developed to detect antibodies directed against or associated with platelet membrane antigens. These assays can be categorized into direct (detecting antibody associated with the patient’s platelets) or indirect (detecting antibody in the patient’s serum that binds to control platelets). Indirect tests may detect, in addition to autoantibodies, alloantibodies (e.g., HLA related), particularly in multiparous women and individuals who have received multiple transfusions, and thus give false-positive results. Autoantibodies tend to be associated with the patient’s platelets and are usually present in low concentration in serum. Therefore a direct test is the preferred test for autoimmune thrombocytopenias such as ITP.
A number of direct assays measure immunoglobulin on platelets, regardless of whether the immunoglobulin is specifically or nonspecifically bound to the platelet surface. These assays generally detect platelet-associated IgG (PAIgG), but they can also be engineered to detect IgM or IgA. PAIgG is increased in immune disorders such as ITP and in nonimmune thrombocytopenic disorders such as leukemia and myelodysplastic syndrome. Thus the specificity of these tests is limited. Moreover, the sensitivity of these tests is limited because autoantibodies, such as those responsible for ITP, represent only a small fraction of the total PAIgG. Megakaryocytes nonspecifically take up plasma proteins, including IgG and albumin, and incorporate these proteins into the alpha granules of platelets, especially in disease states associated with increased thrombopoiesis. Therefore increased PAIgG can result from elevated antibody production for any reason.
More recent assays use antigen capture techniques to detect platelet glycoprotein–specific antibodies, such as those that recognize GPIIb/IIIa and GPIa/IX/V, and have high specificity. Unfortunately, the sensitivity is generally too low for these assays to be used for the routine serologic diagnosis of most immune thrombocytopenias. A newer method is based on flow cytometric detection of autoantibodies reacting with specific platelet proteins immobilized on microbeads.
Macrophage and Platelet Fcγ Receptors.
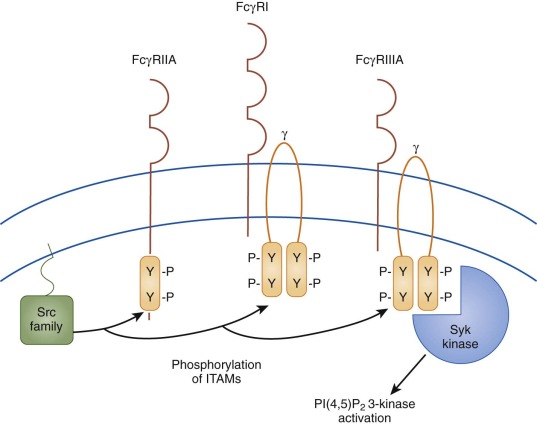
Receptors for the Fc domain of IgG (Fcγ receptors) are found on a variety of cell types, including macrophages and platelets. These receptors have been shown to play a critical role in immune complex–mediated platelet destruction. Fcγ receptors are diverse in structure and function and fall into two major classes: those that activate effector functions, such as phagocytosis or platelet activation, and those that inhibit effector functions.
Activating Fcγ receptors on macrophages include the low-affinity receptors FcγRIIA and FcγRIIIA and the high-affinity receptor FcγRI ( Fig. 34-2 ). As discussed later, cross-linking of the activating receptor FcγRIII promotes phagocytosis of antibody-coated platelets in certain disease states, including ITP. The only Fcγ receptor expressed on platelets is the activating receptor FcγRIIA. Binding of IgG complexes to this receptor results in receptor cross-linking, which in turn initiates platelet aggregation and activation. This process is involved in heparin-induced thrombocytopenia (HIT), the most common drug-induced thrombocytopenia.
Immune Thrombocytopenic Purpura.
ITP is a disorder characterized by accelerated destruction of antibody-sensitized platelets by phagocytic cells, especially those of the spleen. In many affected individuals, inhibition of megakaryopoiesis also contributes to the degree of thrombocytopenia. ITP is the most common autoimmune disorder affecting a blood element. The annual incidence is about 1 in 10,000 children. Two major forms are seen: acute ITP and chronic ITP. Acute ITP is usually a benign, self-limited condition that occurs in young children, typically those younger than 10 years. Often a viral infection or vaccination precedes the onset of acute ITP. In most of these patients, the thrombocytopenia resolves within weeks or a few months of the original manifestation. Chronic ITP is defined arbitrarily as persistence of thrombocytopenia (platelet count <150,000/µL) for longer than 6 months after the initial manifestation, although some hematologists advocate a later cutoff point ( Box 34-2 ).
Controversial Issue 1
What is the definition of chronic ITP? Traditionally, chronic ITP has been defined as thrombocytopenia lasting longer than 6 months. Many pediatric hematologists take exception to this definition because a significant fraction of children with ITP recover in 6 to 12 months. The Intercontinental Childhood ITP Study Group has recommended that a 12-month rather than a 6-month cutoff point be used to define chronicity.
Controversial Issue 2
Is it necessary to perform bone marrow aspiration in patients with suspected ITP before starting glucocorticoid therapy? Although the diagnosis of leukemia is extremely unlikely when the clinical history, physical examination, and peripheral smear are consistent with ITP, many hematologists routinely perform bone marrow aspiration before initiating glucocorticoid therapy. Retrospective studies suggest that this may not be necessary. Bone marrow aspiration or biopsy is warranted for children with atypical laboratory features and for those in whom initial therapy fails.
Controversial Issue 3
Where does splenectomy belong in the therapeutic decision tree for chronic ITP? Experts generally agree that splenectomy should be deferred as long as possible and be reserved for patients with severe, symptomatic thrombocytopenia. The favorable safety profile of rituximab and the possibility of a sustained response could justify its use instead of splenectomy in the decision tree, but this has yet to be demonstrated in a prospective clinical trial. Despite the invasive and irreversible nature of splenectomy, some pediatric hematologists advocate the use of rituximab only after splenectomy has failed because splenectomy has a superior 5-year response rate compared with rituximab (71% versus 26%).
ITP, Immune thrombocytopenic purpura.
Children in whom ITP is diagnosed have an excellent chance of spontaneous recovery, irrespective of therapy. The platelet count returns to normal in 4 to 8 weeks in approximately half of the patients and by 3 months after diagnosis in two thirds of children. In a review of 12 publications involving more than 1500 children with ITP, 76% achieved complete remission within 6 months of initial evaluation. Spontaneous recovery was documented in 37% of the remaining patients with thrombocytopenia persisting longer than 6 months. These findings have been confirmed in other large studies. Factors associated with the development of chronic ITP include age older than 10 years, insidious onset, and female gender.
Pathogenesis.
ITP is caused by autoantibodies that interact with membrane glycoproteins on the surface of platelets and megakaryocytes. These antibodies result in accelerated platelet destruction and may also impair thrombopoiesis. More than a third of adults with ITP have inadequate platelet production despite increased numbers of megakaryocytes in their bone marrow. Certain antiplatelet antibodies have been shown to inhibit megakaryocytopoiesis or egress of platelets from the marrow space. The GPIIb/IIIa complex is the autoantigen implicated most often as the cause of childhood and adult ITP. Autoantibodies directed against the GPIb/IX/V and GPIa/IIa complexes have also been reported. In rare instances platelet glycolipids have been implicated as autoantigen targets in chronic ITP. Autoantibodies diminish or disappear when platelet levels are restored to normal.
Factors that trigger platelet autoantibody formation in acute ITP are not well understood. Although various mechanisms by which viruses may induce autoimmune disease have been suggested, the link between the immune response initiated by infection (or vaccination) and the subsequent production of platelet autoantibodies has not been established. Proposed mechanisms include adsorption of virus to platelets, deposition of virus-containing immune complexes onto platelet membranes, or exposure of cryptic neoantigens on the platelet surface. There are data to both support and refute the hypothesis that acute ITP is triggered by antiviral antibodies that cross-react with platelet antigens.
Some evidence suggests that T lymphocytes also play a role in the pathogenesis of ITP. Dysregulated helper T cells can promote the expansion of antiplatelet antibody–producing B-cell clones. Cytotoxic T cells from some ITP patients have the capacity to destroy platelets ex vivo. CD3+ T lymphocytes from ITP patients are resistant to glucocorticoid-induced apoptosis, thus suggesting that disturbed apoptosis may contribute to defective clearance of autoreactive T lymphocytes.
Genetic factors have been proposed to influence the development of ITP. Studies have failed to detect linkage of the disease to particular HLA genotypes. Polymorphisms in genes encoding phagocyte Fcγ receptors or proinflammatory cytokines may influence the development of ITP. Immunodeficiency states that have been associated with chronic ITP are discussed later.
Clinical and Laboratory Features.
The typical manifestation of acute ITP is the abrupt onset of bruising and bleeding in an otherwise healthy child. Frequently, there is a history of a viral illness in the weeks preceding the onset of bruising. Seasonal fluctuation in the diagonsis of ITP has been noted, with a peak during spring and a nadir in the autumn. Petechiae and ecchymoses are evident in most patients. Epistaxis and oral mucosal bleeding are seen in fewer than a third of patients. Hematuria, hematochezia, or melena is evident in fewer than 10%. Menorrhagia may be observed in adolescent women with ITP. Although children with ITP may have extremely low platelet counts, bleeding episodes are less severe in these patients than in those with hypoproductive thrombocytopenia. This finding has been attributed to enhanced platelet production and young, large, hemostatically effective circulating platelets. A palpable spleen is present in about 10% of reported cases of childhood ITP; this finding alone should not justify performing imaging studies on the affected child’s abdomen. Malaise, bone pain, and adenopathy are uncommon and should raise concern for another cause, such as acute leukemia.
The peak age at diagnosis is 2 to 6 years. Although acute ITP may be diagnosed in children of any age, adolescents and infants are more likely to have chronic ITP develop in combination with some other immune disorder. In children ITP is seen equally in males and females, whereas in adults ITP is seen predominantly in females by a 2-to-1 ratio ( Table 34-1 ).
| Childhood ITP | Adult ITP |
|---|---|
| Females = males | Females > males (2 : 1) |
| Abrupt onset | Insidious onset |
| Infectious prodrome common | Infectious prodrome uncommon |
| <20% chronic | >50% chronic |
Roughly 80% of children have platelet counts below 20,000/µL, often less than 10,000/µL. Leukocyte and red cell counts are usually normal, although anemia may be seen in as many as 15% of these children, especially those with a significant history of epistaxis, hematuria, or gastrointestinal bleeding. A review of the peripheral blood film is mandatory in every child suspected of having ITP; features inconsistent with a diagnosis of ITP should prompt further investigation. Bone marrow aspiration or biopsy reveals normal or increased numbers of megakaryocytes. Increased numbers of eosinophils and their precursors may be noted, although this is not predictive of outcome.
The need for bone marrow examination in children with typical features of acute ITP is a subject of debate. There is a consensus that bone marrow aspiration is not necessary if the initial management is observation alone or administration of intravenous immunoglobulin (IVIG) or anti-D. Controversy exists regarding whether bone marrow aspiration should be performed in all children before glucocorticoid therapy is started to exclude the possibility of acute leukemia (see Box 34-2 ).
Additional tests that may be considered in the initial evaluation and management of suspected ITP include blood group and Coombs test (required if anti-D therapy is contemplated), quantitative immunoglobulin levels (before IVIG therapy), and antinuclear antibody test. Retrospective studies have shown that the antinuclear antibody test is positive in approximately 30% of pediatric patients with otherwise uncomplicated ITP and may predict a subset of patients at risk for the development of further autoimmune symptoms. Tests not considered useful unless specific reasons are identified in the patient’s history and physical examination include screening coagulation tests, liver and renal function tests, serum complement levels, thyroid function tests, and testing for human immunodeficiency virus (HIV) or Helicobacter pylori infection.
Options for the Initial Management of Immune Thrombocytopenic Purpura.
Childhood acute ITP is usually a benign, self-limited disorder that requires minimal or no therapy in the majority of cases. There is no convincing evidence that medical therapy alters the natural history of the disease. Indications for treatment vary among practitioners and are a source of debate. One set of practice guidelines put forth by the American Society of Hematology (ASH) recommends that children with ITP and platelet counts less than 20,000/µL plus significant mucosal membrane bleeding or those with platelet counts less than 10,000/µL and minor purpura be treated with IVIG or a glucocorticoid. However, many pediatric hematologists take exception to this recommendation. In treatment guidelines put forth by other expert panels, a child’s condition, rather than the platelet count, steers management; children with bruising but without mucosal or more severe hemorrhage may be treated by observation alone, irrespective of the platelet count.
At the heart of the treatment debate is the perceived risk for ICH, a rare but potentially life-threatening complication in children with ITP. In a review of 12 case series involving 1293 children, the incidence of ICH was 0.9%. Other surveys suggest that this may be an overestimate and that the true incidence of ICH in children with ITP is between 0.1% and 0.5%. The subgroup of children with ITP who appear to be at greatest risk for ICH are those with platelet counts less than 20,000/µL and additional risk factors such as a history of head trauma, aspirin use, or arteriovenous malformation. However, the platelet count alone has never been shown to predict the severity of bleeding symptoms.
There is no evidence that medical therapy, such as administration of a glucocorticoid or IVIG, reduces the incidence of ICH. Indeed, retrospective studies have shown that ICH may occur despite previous or concomitant therapy with IVIG or a glucocorticoid. The low incidence of ICH in children with ITP precludes a randomized clinical trial to determine whether treatment reduces the risk.
The results of randomized trials for various treatment approaches have been summarized previously. Regardless of whether pharmacologic therapy is used, detailed education and careful follow-up should be provided to the patient and family. The child’s activities should be limited, and aspirin-containing medications should be avoided. Although hospitalization is appropriate for the treatment of a child with a severe bleeding episode, there is no evidence to support routine hospitalization of patients with otherwise uncomplicated ITP.
Observation Only.
For a patient with ITP and only minor purpura, medical therapy may not be necessary. There is uniform consensus that patients with platelet counts greater than 20,000/µL and only minor purpura do not require therapy. As noted earlier, management of patients with platelet counts lower than 20,000/µL and minor bleeding is controversial.
First-Line Medical Therapies.
Glucocorticoids: Glucocorticoids are presumed to act through several mechanisms, including inhibition of both phagocytosis and antibody synthesis, improved platelet production, and increased microvascular endothelial stability. The latter effect may explain why symptomatic bleeding episodes often subside before the platelet count increases.
In randomized trials prednisone therapy has been shown to induce normalization of the platelet count more promptly than placebo does. Although various doses have been used, the traditional glucocorticoid regimen is prednisone at 2 mg/kg/day (maximum of 60 to 80 mg) for approximately 21 days. A regimen of 4 mg/kg/day for 7 days and then tapered to day 21 is equally effective and appears to cause fewer side effects. Prednisone at a dose of 4 mg/kg/day orally for 4 days with no tapering is also effective. An alternative to these regimens is megadose pulse therapy (methylprednisolone, 30 mg/kg/day intravenously or orally for 3 days). Side effects of glucocorticoid therapy include cushingoid facies, weight gain, fluid retention, acne, hyperglycemia, hypertension, moodiness, pseudotumor cerebri, cataracts, growth retardation, avascular necrosis, and osteoporosis.
IVIG: Imbach and colleagues first reported the successful use of IVIG for the treatment of acute ITP in a small series of children. This was followed by many reports that documented the ability of IVIG to cause a rapid increase in the platelet count in patients with acute and chronic ITP.
IVIG slows the clearance of antibody-coated blood cells from the circulation by inhibiting the phagocytic activity of cells of the reticuloendothelial system. Studies in the early 1980s suggested that this effect is mediated primarily through the Fc portion of IgG. Specially treated preparations of intravenous IgG lacking the Fc portion of the molecule were inferior to unmodified IgG at increasing the platelet count in ITP patients. The proposed mechanism for this effect was Fc receptor blockade. Subsequent studies of FcγRIIB-deficient mice suggested that IVIG elicits its effect by activating these inhibitory receptors and not simply by Fc receptor blockade ( Fig. 34-3 ). However, recent publications cast doubt on the conclusion that IVIG acts through activation of FcγRIIB. Although some data support the notion that anti-idiotypic antibodies may also be present in commercial preparations and contribute to the action of IVIG, the clinical importance of this mechanism remains unproved.
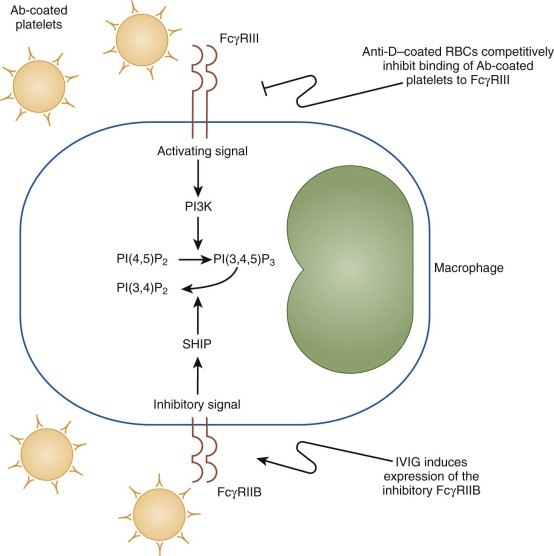
The traditional dose of IVIG is 2 g/kg divided over a period of 2 to 5 days. However, several studies suggest that lower doses are effective. In a randomized trial, pediatric patients who received 0.8 g/kg IVIG had at least as rapid a response rate as those who received 2 g/kg over a 2-day period. In more recent randomized trials, favorable results have been seen with even lower doses of IVIG (250 mg/kg/day for 2 days). Among pediatric patients in whom a response is seen, IVIG produces a more rapid increase in platelet count than do traditional doses of a glucocorticoid (2 mg/kg/day of prednisone), as documented in controlled trials.
Several other general conclusions have emerged from studies on IVIG in pediatric patients. First, both splenectomized and nonsplenectomized patients may respond to IVIG. Second, responses to IVIG are generally reproducible. Third, the duration of the response is brief, approximately 2 to 4 weeks.
Enthusiasm for the use of IVIG is offset by cost considerations. IVIG is considerably more expensive than glucocorticoids or anti-D. One study concluded that IVIG may be cost-effective if it reduces hospital stay or if it prevents the need for a splenectomy.
Complications associated with IVIG are common and occur in 15% to 75% of patients. Frequent side effects include flulike symptoms such as headache, nausea, lightheadedness, and fever. Some of these adverse effects can be alleviated by pretreatment with analgesics or antihistamines. Approximately 10% of patients treated with IVIG (2 g/kg) experience aseptic meningitis manifested as a severe, protracted headache and photophobia. These symptoms, which can cause considerable anxiety in patients, parents, and physicians, may spur additional diagnostic studies (e.g., computed tomography) or result in prolonged hospitalization. A rare but serious side effect of IVIG infusion is anaphylaxis, which can occur in patients who are totally deficient in IgA. Most preparations of IVIG contain small amounts of IgA, and IgE antibodies responsible for anaphylaxis form after an initial exposure to IgA in these preparations.
Anti-D: Anti-D is a plasma-derived immunoglobulin prepared from donors selected for a high titer of anti-Rh 0 (D) antibody. Anti-D elicits a rise in platelet count, along with mild to moderate anemia, in most patients with ITP. A role for anti-D in the treatment of chronic ITP is now well established. Its utility in acute ITP has been studied less extensively, but it is also effective in this clinical setting.
Anti-D can be used to treat only Rh 0 (D)-positive patients with ITP because Rh 0 (D)-negative patients do not show a response. The presumed mechanism of action is phagocytic cell blockade. Patients with intact spleens are more likely to respond to anti-D than splenectomized patients are.
The generally recommended dose is 50 to 75 µg/kg as a short intravenous infusion or subcutaneous injection. Uncontrolled studies have demonstrated that anti-D treatment increases the platelet count in approximately 80% of Rh 0 (D)-positive children. The therapeutic effect of anti-D lasts for 1 to 5 weeks. A controlled trial showed that anti-D therapy was less effective than IVIG or a glucocorticoid in terms of days required to attain a platelet count of 20,000/µL. Anti-D is less expensive than IVIG, with an estimated cost savings of 35% per episode of ITP. The shorter administration time required for anti-D therapy also makes the medication more convenient to use than IVIG.
Adverse reactions of headache, nausea, chills, dizziness, and fever have been reported in 3% of infusions, and these adverse reactions were classified as severe in only a minority of cases. Some degree of hemolysis, the main adverse reaction with anti-D, is inevitable because of binding of anti-D antibody to Rh 0 (D)-positive erythrocytes. There is laboratory evidence of hemolysis in most patients. The average decline in hemoglobin ranges from 0.5 to 1 g/dL, and most cases of hemolysis do not require medical intervention. In a small subset of patients, more significant hemolysis has been observed, which has tempered enthusiasm for use of the drug. Postmarketing surveillance by the U.S. Food and Drug Administration documented 15 cases of hemoglobinemia or hemoglobinuria after anti-D therapy. Of these patients six required transfusion, eight experienced an onset or exacerbation of renal insufficiency, and two underwent dialysis. The incidence of intravascular hemolysis was estimated to range from 0.1% to 1.5%. Why certain patients experience severe intravascular hemolysis after anti-D therapy is unclear. Subcutaneous delivery of anti-D may be associated with a lower incidence of severe hemolytic reactions.
Relapses or Treatment Failures.
Many patients treated with a glucocorticoid, IVIG, or anti-D will become thrombocytopenic again after a few weeks. These patients are likely to respond again to the therapy used initially. Patients who require therapy and whose thrombocytopenia does not respond to initial treatment with a glucocorticoid, IVIG, or anti-D are usually treated with one of the alternative frontline therapies. No clinical studies document the response rate in these instances. Patients with persistent mild hemorrhage may benefit from low-dose glucocorticoid therapy.
Management of Chronic Immune Thrombocytopenic Purpura.
In approximately 20% of children in whom ITP is diagnosed, thrombocytopenia persists for longer than 6 months. Chronic ITP is more common in female adolescents and adults than in their male counterparts. Chronic ITP sometimes occurs in association with other autoimmune diseases or in conjunction with an underlying condition known to predispose to disorders of autoimmunity, such as lymphoma ( Box 34-3 ).
Immunodeficiency
Hypogammaglobulinemia
Common variable immunodeficiency
Lymphoproliferative disorders
Autoimmune lymphoproliferative syndrome
Hodgkin lymphoma
Collagen vascular disorders
Systemic lupus erythematosus
Infection
Human immunodeficiency virus
In up to a third of children with chronic ITP, spontaneous remission will occur months or years later. It is unclear why the condition resolves in these individuals. An estimated 5% of children with chronic ITP have recurrent ITP characterized by intermittent episodes of thrombocytopenia followed by lengthy periods of remission. This is presumed to reflect a chronic compensated state of ITP. During periods of remission, increased platelet production balances the increased rate of platelet destruction. During exacerbations, platelet production by the marrow is suppressed by viral infections or other factors and is unable to offset the rate of destruction.
Evaluation of patients with chronic ITP should include bone marrow aspiration, if not already done; screening tests for immunodeficiency (e.g., quantitative immunoglobulin levels/subsets, specific antibody titers, lymphocyte subsets), systemic lupus erythematosus (SLE), and other autoimmune diseases (antinuclear antibody tests, direct Coombs test, antiphospholipid antibody tests, thyroid function tests); and serologic testing for HIV. Obtaining blood counts and peripheral blood smears from the parents may help exclude familial disorders.
The primary goal of treatment in patients with chronic ITP is to prevent bleeding, not cure the disease. As with acute ITP, therapy decisions should be based more on symptoms than on platelet count. In formulating a treatment plan, the physician must balance the potential side effects of treatment with the risk of bleeding episodes caused by the disease. Because the goal of treatment is a safe platelet count, not necessarily a normal platelet count, observation alone is an appropriate approach for many patients, especially those with minimal symptoms.
Splenectomy.
Splenectomy is thought to be effective because in most patients the spleen is the major site of both platelet destruction and autoantibody production. Platelet survival studies indicate improved platelet life span after splenectomy.
Splenectomy remains the most predictable intervention to achieve long-term remission in children with chronic ITP. Splenectomy should be considered in a pediatric patient with chronic ITP whose risk for hemorrhage precludes the use of observation alone. However, there is debate about where splenectomy belongs in the therapeutic decision tree for chronic ITP (see Box 34-2 ). Because remissions are often delayed in children and the risk of postsplenectomy sepsis is significant, especially in children younger than 5 years, splenectomy is deferred longer in children with ITP than in adults. ASH practice guidelines recommend that splenectomy be considered for children in whom ITP has persisted for at least 1 year and who have bleeding symptoms and a platelet count of less than 10,000/µL (ages 3 to 12) or 10,000 to 30,000/µL (ages 8 to 12 years). Practice guidelines in the United Kingdom are similar.
Laparoscopic splenectomy is preferred over open splenectomy in children with chronic ITP. Advantages of the laparoscopic procedure include less postoperative pain, earlier return of gastrointestinal function, shorter hospitalization, more rapid resumption of normal activities, and smaller incisions. To ensure adequate hemostasis, the platelet count can be raised preoperatively with a glucocorticoid, IVIG, or anti-D. Despite these interventions, many patients will still have thrombocytopenia at the time of splenectomy; however, most will not exhibit excess bleeding. Accordingly, prophylactic platelet transfusions are not warranted. Instead, platelet transfusions should be reserved for patients with intraoperative bleeding. Most experienced surgeons have become comfortable performing laparoscopic splenectomy on patients with platelet counts in the range of 50,000 to 100,000/µL. Overall perioperative mortality is less than 1%.
The majority of children with chronic ITP respond to splenectomy. In a review of 16 case series involving 271 children undergoing elective splenectomy for chronic ITP, a complete remission rate of 72% was reported. The platelet count usually rises immediately after splenectomy and reaches a maximum 1 to 2 weeks postoperatively. If the peak platelet count achieved after splenectomy is greater than 500,000/µL, permanent remission is likely. Certain patient characteristics are associated with a higher probability of response to splenectomy, including a previous response to glucocorticoid therapy. However, there is no single factor or combination of factors that will predict the response to splenectomy in all cases.
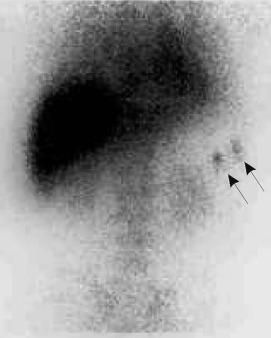
Relapse after an immediate response to splenectomy may result from transient viral suppression of thrombopoiesis. If the thrombocytopenia persists, an accessory spleen should be considered. Approximately 40% of patients with persistent thrombocytopenia will have a residual accessory spleen that can be seen with sensitive imaging techniques such as 99m Tc sulfur colloid scans ( Fig. 34-4 ) or In platelet localization studies. Many of these patients will achieve complete remission after removal of the accessory spleen. The presence of Howell-Jolly bodies on a peripheral blood smear does not rule out an accessory spleen.
The major risk after splenectomy is fatal sepsis caused by encapsulated organisms. The risk of septicemia in splenectomized patients is estimated to be 1 per 300 to 1000 patient-years. To lessen the risk of future sepsis, patients should receive vaccinations for pneumococcus, Haemophilus influenzae type b, and meningococcus at least 2 weeks before surgery. In addition, penicillin prophylaxis is indicated for splenectomized patients younger than 5 years. The benefit of penicillin prophylaxis in older asplenic children is controversial.
Phagocyte Blockade.
Intermittent maintenance therapy with a glucocorticoid, IVIG, or anti-D can be used to delay splenectomy. These medications may also be useful in the management of patients with chronic ITP who experience symptomatic thrombocytopenia after splenectomy.
In some patients a safe platelet count can be maintained with low doses of prednisone. However, even low doses of prednisone, comparable to physiologic cortisol secretion, can cause osteoporosis.
IVIG (1 to 2 g/kg total dose administered) elevates the platelet count in most children with chronic ITP. Periodic doses of IVIG may be used as maintenance therapy to defer splenectomy in young children with chronic ITP. This approach is cost-effective in young children. In approximately 25% of children, ITP will become refractory to maintenance therapy with IVIG. Low-dose alternate-day prednisone therapy can be used as an adjunct to maintenance IVIG therapy. Although repeated infusions of IVIG induce short-term platelet responses in most patients with chronic ITP, there is no evidence that such therapy will affect the natural history of the disease and induce a long-lasting remission.
Anti-D administration raises the platelet count in most Rh 0 (D)-positive children with chronic ITP. However, the benefit is usually transient, with a median duration of 3 to 5 weeks. Anti-D can be effective as maintenance therapy in patients who are not appropriate candidates for splenectomy or other therapies because of young age or other factors. In Rh 0 (D)-positive patients with chronic ITP, anti-D is preferred over IVIG because of ease of administration, comparable efficacy, and lower cost.
Monoclonal Antibodies That Target B Cells or T Cells.
Rituximab: Rituximab, a humanized monoclonal antibody directed against CD20, is now commonly used to treat chronic ITP despite limited clinical trial data documenting its efficacy in children with this condition (see Box 34-2 ). Rituximab is generally administered in four weekly intravenous infusions of 375 mg/m 2 each, but there is no standard dose for rituximab in children. There are reports of children with ITP who responded to lower doses of rituximab. This agent causes rapid depletion of pre-B cells and B cells that lasts for 6 to 12 months. Pre-B cells efficiently present antigen to T cells, and disruption of this interaction may attenuate the autoimmune response. Approximately 30% to 60% of patients with chronic ITP respond to rituximab. The initial response may be immediate (within 1 week) or delayed (up to 3 months). Relapses are common but often respond to retreatment. The estimated 5-year sustained response rate in children is 26%. Side effects of rituximab include fever, urticaria, pruritus, throat tightness, and serum sickness. Another serious complication associated with rituximab is progressive multifocal leukoencephalopathy (PML). More than 50 cases of PML have been reported in patients treated with rituximab, including a patient with ITP.
Alemtuzumab: Alemtuzumab (Campath 1H) is a humanized monoclonal antibody that targets CD52, a protein on both B and T cells. There are anecdotal reports of its use in patients with chronic ITP. Part of the effect of alemtuzumab can be attributed to B-cell depletion, analogous to rituximab, and part to depletion of T cells. Unlike rituximab, alemtuzumab induces profound generalized immunosuppression associated with opportunistic infections.
Other Immunosuppressive Agents.
A large number of other immunosuppressive medications have been used to treat refractory ITP, as reviewed elsewhere. No randomized studies have evaluated the effectiveness of these various therapies. The choice of therapy must be individualized to the patient.
Azathioprine: Azathioprine acts preferentially on lymphocytes and was one of the first drugs reported to be effective in the treatment of chronic refractory ITP. Recommended doses range from 50 to 200 mg/m 2 /day orally. Continuous therapy for 4 to 6 months is required before a patient is considered unresponsive. Once a response is obtained, the dose should be tapered to the lowest level that results in a hemostatic platelet count. In adults with refractory ITP treated with azathioprine, approximately 20% achieve a complete response and 45% exhibit a partial response. Relapse often occurs when the therapy is stopped. The combination of azathioprine and oral steroids may be synergistic, and azathioprine therapy may allow a reduction in the dose of glucocorticoids. Toxicities include dose-related leukopenia, opportunistic infection, and an increased risk for malignancy.
Cyclophosphamide: The alkylating agent cyclophosphamide has also been used to treat chronic refractory ITP, with a response rate similar to that with azathioprine. Greater risks are associated with the use of cyclophosphamide than with azathioprine. Side effects include myelosuppression, alopecia, nausea, infertility, teratogenicity, hemorrhagic cystitis, and an increased risk for malignancy.
Cyclosporine: The potent immunosuppressive agent cyclosporine disrupts T-cell function through calcineurin inhibition. There are case reports of patients with refractory ITP who had increases in platelet counts after treatment with cyclosporine. Response rates vary. A starting dose of 5 mg/kg/day given in divided oral doses is recommended, with the aim of maintaining a serum cyclosporine level of 200 to 400 ng/mL. The drug should be discontinued after 4 weeks if there is no response. Hypertension, renal insufficiency, hirsutism, and liver dysfunction are common side effects.
Tacrolimus: Though not structurally related to cyclosporine, tacrolimus has a similar mechanism of action and has been used anectodally to treat chronic ITP. Initial dosing recommendations for tacrolimus in children are 0.15 to 0.3 mg/kg/day given in divided oral doses. Doses should be adjusted to maintain trough tacrolimus concentrations of 5 to 20 ng/mL in whole blood. Common adverse effects associated with tacrolimus include hypertension, tremor, headache, and hyperglycemia.
Mycophenolate mofetil: There are reports showing the effectiveness of another immunomodulatory drug, mycophenolate mofetil (MMF), in patients with steroid-resistant chronic ITP. MMF is a prodrug of mycophenolic acid, a noncompetitive inhibitor of inosine-5α-monophosphate dehydrogenase, a key enzyme in the purine biosynthetic pathway. MMF inhibits proliferation of both T cells and B cells. In one series a sustained platelet increase to a level greater than 50,000/µL was seen in 7 of 18 patients with refractory ITP. For adults the usual starting dose is 250 mg orally twice daily, with an increase to 1 g twice daily by 3 weeks. MMF has few side effects and is generally well tolerated.
Vinca alkaloids: The vinca alkaloids vincristine and vinblastine may induce a response in some patients with chronic refractory ITP. These drugs bind tubulin and inhibit microtubule polymerization, which is presumed to disrupt phagocytosis. Either vincristine or vinblastine may be used; the efficacy of the two drugs is comparable. The usual dose of vincristine is 1.5 mg/m 2 (maximum of 2 mg) intravenously, whereas the dose of vinblastine is 6 mg/m 2 (maximum of 10 mg) intravenously. These doses are repeated weekly for a month. If no response is seen, additional doses are unwarranted because subsequent improvement is unlikely. Patients who show a response often require repeated doses at 2- to 3-week intervals to maintain a safe platelet count. In adults the response rate is approximately 12%, and the partial response rate is 35%. The response rate in children is not established and varies considerably in the reports available. Adverse effects include peripheral neuropathy, constipation, and alopecia. Vinblastine causes dose-related myelosuppression.
Danazol: Danazol, an attenuated androgen with mild virilizing effects, has been shown to increase the platelet count in patients with refractory ITP, although the mechanism of action is uncertain. Approximately 40% of adult patients respond to danazol with an increase in platelet count to greater than 50,000/µL. A typical dose is 300 to 400 mg/m 2 /day orally, although smaller doses (50 mg/m 2 ) have been reported to be effective. In general, 2 months of therapy is required before a response is seen. Often the medication must be continued to maintain the platelet count, although effort should be made to minimize the dose. Danazol may be especially useful for the treatment of older adolescent or adult women with chronic, refractory ITP and uncontrollable menorrhagia. Side effects of danazol include acne, fluid retention, hirsutism, deepening of the voice in women, headache, nausea, rash, breast tenderness, and oligomenorrhea or amenorrhea. Patients should be monitored for the development of liver toxicity.
Interferon alfa: Although the mechanism is unknown, 25% of adults with chronic ITP treated with interferon alfa display a significant, albeit transient, increase in platelet count. Interferon-alfa has also been used to treat children with chronic ITP. The recommended dose is 3 million U/m 2 subcutaneously three times per week for 4 weeks. Adverse effects of interferon alfa include flulike symptoms and neutropenia.
Combination chemotherapy: Combination chemotherapy with or without autologous stem cell transplantation has been used successfully to treat some adults with chronic, refractory ITP, with an overall sustained remission rate of approximately 40%.
Thrombopoiesis-Stimulating Agents.
In ITP the rate of platelet production is inadequate to offset the increased rate of platelet destruction. Two newer drugs for the treatment of chronic ITP act by stimulating thrombopoiesis. Romiplostim is a recombinant protein that contains two domains: a peptide that binds to the thrombopoietin (TPO) receptor c-Mpl and an antibody Fc domain that increases the half-life in circulation. In a placebo-controlled, phase II study this agent was efficacious when administered subcutaneously on a weekly basis (1 to 3 µg/kg) to adults with chronic ITP. Toxicity was minimal. In children there has been one randomized clinical trial of romiplostim and several case series. Romiplostim increased the platelet count in 88% of the children in this study, and quality-of-life measurements documented reduced parental burden. Another small-molecule c-Mpl agonist for chronic ITP is eltrombopag, which binds to the transmembrane region of c-Mpl rather than the TPO binding site. This agent is given orally once daily. A phase II trial on adults with chronic ITP showed that eltrombopag at doses of 50 and 75 mg was significantly better than placebo at increasing platelet counts to greater than 50,000/µL. The drug appears to have few side effects, one of which is reversible liver toxicity. Romiplostim and eltrombopag are licensed for use in adults with chronic, severe ITP but not yet for use in children.
One concern is that myelofibrosis may develop after prolonged treatment with TPO receptor agonists. A slight but significant increase in reticulin fibrosis has been demonstrated in the bone marrow of adult patients receiving TPO receptor agonists. Until additional safety data are collected, monitoring for marrow changes with an annual bone marrow biopsy seems prudent. Thromboembolic events have also been reported in patients treated with romiplostim and eltrombopag. Other limitations for the use of TPO receptor agonists include cost, the indefinite duration of therapy required, and the risk of severe thrombocytopenia in patients who interrupt therapy.
Management of Life-Threatening Hemorrhage in Immune Thrombocytopenic Purpura.
ICH or another life-threatening form of bleeding mandates intervention with multiple therapeutic modalities, including IVIG (1 g/kg), anti-D (50 to 75 µg/kg), high-dose glucocorticoids (e.g., methylprednisolone, 30 mg/kg intravenously), platelet transfusions, or a combination of these interventions. Because IVIG and anti-D act to inhibit phagocytosis by distinct and potentially synergistic mechanisms (see Fig. 34-3 ), these two agents may be used together for instances of severe bleeding. TPO receptor agonists have a limited role in this setting because it takes an average of 2 weeks to see a response. Recombinant factor VIIa has also been used in the management of ICH in patients with thrombocytopenia refractory to platelet-enhancing agents. If a craniotomy is required, consideration should be given to splenic artery embolization or emergency splenectomy, although most hematologists reserve splenectomy for patients in whom ITP does not respond rapidly to therapy with glucocorticoids, IVIG, anti-D, and platelet transfusions. Optimal therapy for ICH also includes supportive care with mechanical ventilation and mannitol.
Thrombocytopenia Associated with Lymphoproliferative and Autoimmune Disorders.
Chronic ITP may be a manifestation of an associated autoimmune disease or immunodeficiency state. This is especially true in young children in whom chronic, refractory ITP develops or in adolescent girls with systemic symptoms.
Autoimmune lymphoproliferative syndrome (ALPS) is a rare syndrome that occurs in early childhood and is characterized by massive, nonmalignant lymphadenopathy, splenomegaly, hypergammaglobulinemia, and autoimmune manifestations, including immune thrombocytopenia and hemolytic anemia (see Chapter 13 ). ALPS has been linked to inherited defects in FAS and other genes that regulate lymphocyte apoptosis. An abnormal accumulation of lymphocytes results in lymphadenopathy, hepatosplenomegaly, and hypersplenism. In young children the lymphadenopathy may be pronounced and then wane later in life. Impaired elimination of autoreactive lymphocytes also causes autoimmune manifestations, including autoantibody formation. Patients with ALPS commonly exhibit episodic autoimmune cytopenia, including thrombocytopenia, hemolytic anemia, neutropenia, or combined cytopenias.
A significant proportion of young children with chronic ITP may have a variant of ALPS. For example, in one survey of 20 pediatric patients with chronic cytopenia, 25% were found to have profound defects in Fas-mediated lymphocyte death. Two of these patients, ages 4 and 9 years, had chronic refractory ITP without other clinical manifestations of ALPS. Neither patient had an identifiable mutation in FAS or caspase – 10, and it is presumed that they may have defects in one of the molecules involved in Fas signaling. Similar results have been reported in other studies, thus suggesting that altered Fas signaling, independent of FAS mutations, is associated with and may precipitate chronic hematologic autoimmunity, including ITP.
The lymphadenopathy and cytopenias associated with ALPS generally respond to glucocorticoid therapy. Mycophenolate mofetil and sirolimus are also highly effective in the treatment of ALPS. Because rituximab may lead to prolonged hypogammaglobulinemia when used in ALPS, alternative immunosuppressants should be tried before using this agent. Patients with ALPS have a very high risk of postsplenectomy sepsis, so splenectomy is not recommended.
Thrombocytopenia occurs at some time in approximately 25% of patients with SLE and may be the first manifestation of autoimmunity. Both autoantibodies and complement-fixing immune complexes have been implicated in the pathogenesis of thrombocytopenia in SLE. In addition, antiphospholipid antibodies are frequently observed in the sera of patients with SLE. Antibodies against antigens nonspecifically adsorbed to the surface of platelets, such as DNA, have also been reported.
The initial therapy for thrombocytopenia in patients with SLE is administration of a glucocorticoid, but there is a high incidence of recurrent thrombocytopenia when the steroid dose is tapered. Long-term use of low doses of glucocorticoids may be justified, especially if these drugs also control the other symptoms of SLE. Splenectomy can be used in the management of patients with SLE and chronic, symptomatic thrombocytopenia. In some case series splenectomy for SLE has been shown to yield response rates comparable to those for ITP, whereas in other series the response rate has been lower.
Thrombocytopenia may be associated with an antiphospholipid antibody syndrome, whether the syndrome occurs as a primary disorder or in association with SLE or another autoimmune disorder. Patients with antiphospholipid antibody syndrome often experience recurrent venous or arterial thrombotic complications rather than bleeding (see Chapter 35 ). Autoantibodies against GPIIb/IIIa, GPIa/IIa, and GPIb/IX have been documented in this syndrome. Enhancement of platelet activation by antiphospholipid antibodies may contribute to the prothrombotic state in these patients. In general, the degree of thrombocytopenia is moderate. A variety of therapies have been used to treat antiphospholipid antibody syndrome and the accompanying thrombocytopenia, including glucocorticoids and other immunosuppressive agents.
Evans syndrome is the coexistence of autoimmune hemolytic anemia and thrombocytopenia. Autoimmune neutropenia also develops in many of these patients (see Chapter 13 ). Frequently, patients with Evans syndrome have associated disorders, such as dysgammaglobulinemia. Distinct antiplatelet and antierythrocyte antibodies rather than cross-reactive antibodies are responsible for the association of autoimmune hemolytic anemia and thrombocytopenia. In many patients the course of the disorder is marked by exacerbations of thrombocytopenia, anemia, or neutropenia. Unlike the majority of patients with ITP or isolated autoimmune hemolytic anemia, therapy with glucocorticoids, IVIG, or splenectomy often fails in patients with Evans syndrome. Patients with Evans syndrome have been treated with rituximab, which induces remission in the majority, although relapses tend to occur within 12 months. Other options for treatment include mycophenolate mofetil and cyclosporine. For severe and refractory cases, hematopoietic stem cell transplantation is a consideration.
Immune thrombocytopenia can complicate other autoimmune diseases, including dermatomyositis, juvenile rheumatoid arthritis, Hashimoto thyroiditis, Graves disease, myasthenia gravis, inflammatory bowel disease, sarcoidosis, and protein-losing enteropathy. Immune thrombocytopenia can also precede, accompany, or follow neoplastic disease, especially lymphoproliferative disorders such as Hodgkin and non-Hodgkin lymphoma. Immune thrombocytopenia has also been seen in association with childhood acute lymphoblastic leukemia. In some patients the thrombocytopenia of neoplastic disease resolves in response to antineoplastic therapy, whereas in others the thrombocytopenia requires specific treatment.
Thrombocytopenia occurs in 5% to 10% of patients with HIV infection. Immune-mediated platelet destruction is a major contributor to the thrombocytopenia seen in patients with HIV. The antibodies in HIV-associated thrombocytopenia are directed against the same target epitopes commonly seen in idiopathic ITP—namely, epitopes on GPIIb/IIIa. Immune complexes composed of anti-idiotype antibodies and antibodies directed against the HIV antigen gp160/120 may also induce platelet destruction. Whereas half of patients with ITP have normal or increased platelet production, most patients with HIV-associated thrombocytopenia have decreased platelet production. Infection of megakaryocytes or bone marrow stromal cells may account for the reduced platelet production noted in HIV-associated thrombocytopenia.
Thrombocytopenia may occur early in the course of HIV infection and is often seen within the first 2 years. Platelet counts are rarely lower than 50,000/µL, and symptomatic bleeding is uncommon. However, in HIV-infected patients with hemophilia, the bleeding can be more severe, even life threatening. Therapy is similar to that for ITP, except that HIV-associated thrombocytopenia may respond to antiviral therapy. Patients with severe or symptomatic thrombocytopenia that persists despite antiviral therapy should be managed in the same way as patients with severe ITP.
Neonatal Alloimmune Thrombocytopenia.
NAIT, also termed fetomaternal alloimmune thrombocytopenia, is a syndrome characterized by transient, isolated, severe thrombocytopenia. The syndrome results from placental transfer of maternal alloantibodies directed against paternally inherited antigens present on fetal platelets but absent from maternal platelets. Therefore this condition is the platelet counterpart of hemolytic disease of the newborn.
Alloantigens.
Platelet alloantigens and the platelet membrane glycoproteins on which they reside are listed in Table 34-2 . Most of the antigens were originally named on the basis of the patient’s surname. In many cases duplicate names were assigned to the same alloantigens (e.g., Pl A1 = Zw a , Pen a = Yuk b ). The HPA, or human platelet antigen, nomenclature was proposed in the 1990s to systemize platelet alloantigens. Each HPA system represents a biallelic polymorphism caused by a single amino acid substitution (see Table 33-2 ).
| Current Designation | Original Designation | Platelet Localization | Polymorphism | PHENOTYPE FREQUENCY | |
|---|---|---|---|---|---|
| Whites | Japanese | ||||
| HPA-1a | Pl A1 , Zw a | GPIIIa | Leu33 | 0.98 | 0.99 |
| -1b | Pl A2 , Zw b | Pro33 | 0.027 | 0.037 | |
| HPA-2a | Ko b | GPIb α | Thr145 | 0.99 | ND |
| -2b | Ko a , Sib a | Met145 | 0.017 | 0.25 | |
| HPA-3a | Bak a , Lek a | GPIIb α | Ile843 | 0.85 | 0.79 |
| -3b | Bak b , Lek b | Ser843 | 0.66 | 0.71 | |
| HPA-4a | Pen a , Yuk b | GPIIIa | Arg143 | >0.999 | >0.999 |
| -4b | Pen b , Yuk a | Gln143 | <0.0001 | 0.017 | |
| HPA-5a | Br b , Zav b | GPIa | Glu505 | 0.99 | 0.99 |
| -5b | Br a , Zav a | Lys505 | 0.021 | 0.087 | |
| HPA-6b | Tu a , Cu | GPIIIa | Gln489 | <0.01 | ND |
| HPA-7b | Mo a | GPIIIa | Pro407 | <0.01 | ND |
| HPA-8b | Sr a | GPIIIa | Cys636 | <0.01 | ND |
* Other low-frequency human platelet alloantigens exist but are not listed here. Comprehensive reviews are available.
In white populations the most frequently implicated alloantigen in NAIT is HPA-1a (Pl A1 ), which accounts for 78% of serologically confirmed cases. Incompatibility for the HPA-5b alloantigen is the next most common cause. Approximately 98% of white individuals express the HPA-1a antigen on their platelets; most of the remaining 2% are homozygous for an alternative allele that encodes the HPA-1b (Pl A2 ) antigen. The protein products of these two alleles differ only by a single amino acid (Leu33 versus Pro33). The incidence of NAIT is approximately 1 in 2000 births, which is much lower than the predicted incidence based on distribution of the HPA-1a and HPA-1b alleles in the population. Other immune response genes appear to regulate HPA-1a alloantibody formation. Women with the HLA-DR3 antigen have about a twentyfold relative risk of forming HPA-1a alloantibodies. In Asian populations incompatibility of the HPA-4 system is the most common cause of NAIT (80%), followed by incompatibility of the HPA-3 system. HLA class I antigens are expressed on platelets, and maternal sensitization to HLA has been implicated as a cause of NAIT in a few patients. HLA alloantibodies are present in 10% to 30% of pregnant women, especially multiparous women. The relative rarity of NAIT secondary to HLA alloantibodies may result from binding of these antibodies to the placenta or other tissues. Blood group ABH alloantibodies have been found in a small number of patients with NAIT.
Clinical and Laboratory Features.
The degree of thrombocytopenia in NAIT can be severe, with platelet counts of less than 10,000/µL on the first day of life. Common hemorrhagic manifestations include petechiae (90%), hematomas (66%), and gastrointestinal bleeding (30%). The maternal platelet count is normal, an important laboratory value for distinguishing this condition from neonatal thrombocytopenia secondary to maternal ITP (discussed later). Unlike Rh disease of the newborn, NAIT can occur in both the first and subsequent pregnancies. A history of a previously affected infant provides strong supportive evidence for a diagnosis of NAIT.
Patients with NAIT are at increased risk for ICH, both prenatally and postnatally. About 15% of affected neonates have ICH, and 50% of these cases occur antenatally. In utero bleeding can result in hydrocephalus, porencephaly, seizures, or fetal loss. The pronounced bleeding diathesis associated with anti–HPA-1a alloimmune thrombocytopenia may reflect the combination of severe thrombocytopenia and antibody-mediated interference with the function of the GPIIb/IIIa complex on the few remaining platelets.
Treatment.
The diagnosis of NAIT is confirmed by serologic or genotypic testing, including immunophenotyping of maternal, paternal, and occasionally neonatal platelets; maternal or fetal serum also can be examined for the presence of antiplatelet antibody. Several sensitive assays have been developed for platelet antigen typing and detection of alloantibody. Despite the availability of sensitive tests, serologic evidence of alloantibodies is lacking in a least a third of cases identified by clinical criteria; therefore NAIT is a clinical diagnosis. Because delays may be associated with serologic testing, therapy should be initiated as soon as the diagnosis is suspected.
The treatment of choice for severe NAIT (platelet count <30,000/µL or clinically significant bleeding) is transfusion of washed, irradiated maternal platelets ( Fig. 34-5 ). Irradiation prevents transfusion-associated graft-versus-host disease. In the interest of time, normal donor screening procedures may have to be abridged. There are often medical or practical reasons that maternal platelets cannot be collected in a timely manner. Random-donor platelet infusions can be used in the treatment of NAIT if maternal platelets are not immediately available. However, only a modest and short-lived increase in the platelet count may be observed after the infusion of random-donor platelets. IVIG (1 g/kg daily for 2 days) or a glucocorticoid (methylprednisolone, 2 mg/kg/day) can also be used as a temporary measure, although these interventions may be of limited benefit. If the offending alloantigen is known, platelets from an antigen-negative unrelated donor may be used. In white populations the use of HPA-1a– and HPA-5b–negative donors has been advocated because these two antigens are responsible for more than 95% of cases of NAIT. To provide transfusion support for fetuses and neonates with NAIT, the National Blood Service of England has established an accredited platelet donor panel of HPA-1a–negative individuals with no antibodies to HLA, HPA, or red cell antigens. The majority of these donors are HPA-5b negative.
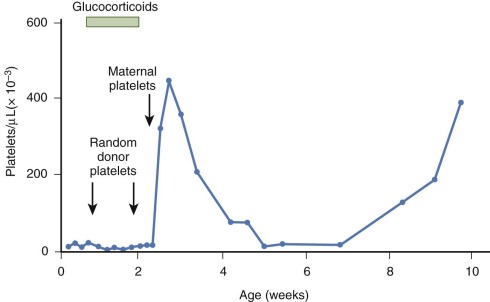
Platelets produced by neonates with alloimmune thrombocytopenia may exhibit accelerated clearance for weeks, until antiplatelet antibody is removed from the circulation (see Fig. 34-5 ). Consequently, it is not unusual for additional transfusions of maternal (or an antigen-compatible unrelated donor) platelets to be required after the first few weeks of life.
Management of Future Pregnancies.
The likelihood is high that subsequent infants born to mothers of infants with NAIT will be affected. Moreover, the severity of antenatal and postnatal hemorrhage tends to increase in later pregnancies. Therefore detailed counseling on the risk of recurrence should be provided to the family at the time that thrombocytopenia is diagnosed in the first infant. Although the neonate’s thrombocytopenia may have resolved before completion of the full diagnostic laboratory evaluation, it is important that the diagnosis and presumed antigen incompatibility be confirmed by formal serologic and genotypic testing to guide the management of future pregnancies. Antibody titers begin to decline after delivery; thus the evaluation should ideally be initiated in the postpartum period. Because the likelihood of thrombocytopenia in subsequent infants depends on whether the father is HPA-1a homozygous or HPA-1a/1b heterozygous, performance of DNA-based genotyping is desirable.
Fetuses at risk for ICH should be monitored with serial ultrasound examinations, although fetal ICH may not always be detected by this method. In some centers prenatal diagnosis of the platelet alloantigen genotype has been performed with DNA obtained by chorionic villus sampling or amniocentesis. More commonly, percutaneous umbilical blood sampling is performed at 20 to 22 weeks of gestation to obtain a fetal platelet count and perform platelet antigen typing. However, this procedure may be associated with fetal loss because of exsanguination; thus compatible antigen-negative (e.g., maternal) platelets should be available when percutaneous umbilical blood sampling is performed and should be infused if the fetus is severely thrombocytopenic.
When the diagnosis of alloimmune thrombocytopenia is confirmed in the fetus, two antenatal treatment approaches are available. In most North American centers, the favored approach is weekly maternal infusion of IVIG. In some European centers the preferred approach for severe cases of alloimmune thrombocytopenia is repeated in utero platelet transfusions; administration of IVIG and a glucocorticoid is reserved for mildly affected fetuses.
Neonatal Autoimmune Thrombocytopenia.
This condition can occur in infants of mothers with immune thrombocytopenia as a result of ITP, SLE, or other autoimmune disorders and is caused by placental transfer of maternal autoantibodies. Antigens implicated as causes of neonatal autoimmune thrombocytopenia mirror those seen in ITP. In general, this condition is less serious than NAIT.
This diagnosis should be considered when both the neonate and the mother exhibit signs of thrombocytopenia or in infants of mothers with a previous history of immune thrombocytopenia. Neonatal thrombocytopenia has been observed in infants of mothers rendered asymptomatic for chronic ITP by splenectomy. Maternal ITP must be distinguished from the mild decrease in platelets that frequently accompanies pregnancy at term, presumably because of plasma volume expansion.
The thrombocytopenia associated with maternal immune thrombocytopenia is generally milder than NAIT. The incidence of cord platelet counts lower than 50,000/µL is only about 3%, and the incidence of ICH is only about 1%. Although clinically significant bleeding episodes are rare, newborns with this condition should be monitored closely because platelet counts often decrease in the days after birth. The threshold for medical intervention in neonates with thrombocytopenia differs from that in older children with ITP because of concern about ICH in the newborn. A platelet count of less than 40,000 to 50,000/µL in a neonate is considered by many to be a useful indicator for starting therapy. IVIG (1 g/kg daily for 2 days) elevates the platelet count in most patients. A glucocorticoid (methylprednisolone, 2 mg/kg/day) may be used in addition to or in lieu of IVIG therapy.
Perinatal management of mothers with immune thrombocytopenia, including the mode of delivery of a thrombocytopenic fetus, is controversial. Because the cord platelet count rarely is below 50,000/µL, perinatal ICH is uncommon and not necessarily related to delivery. The degree of maternal thrombocytopenia is a poor predictor of the degree of neonatal thrombocytopenia, although the clinical course of thrombocytopenia in the first sibling does predict that of the next sibling. Otherwise, there are no reliable predictors of severe thrombocytopenia in the newborn. Direct determination of the fetal platelet count by percutaneous umbilical venous sampling can be performed, but this procedure is rarely justified for this condition.
Posttransfusion Purpura.
PTP is a very rare syndrome in childhood. Typically, this syndrome affects a multiparious woman with HPA-1a–negative platelets who is reimmunized with that alloantigen through blood transfusion. Severe, acute thrombocytopenia ensues 1 week later, presumably reflecting an anamnestic response to the platelet alloantigen. PTP has been reported to develop in males and in nulligravida females as a result of initial exposure to the alloantigen through a previous blood transfusion. HPA-1a is the alloantigen found in 90% of occurrences of PTP.
Alloantibodies cause PTP, but the mechanism of platelet destruction is unclear. Several hypotheses have been put forward to explain how alloantibody promotes destruction of autologous platelets, including immune complex–mediated platelet destruction; adsorption of allogeneic platelet antigens onto autologous platelets, which renders them susceptible to alloimmune destruction; and transient induction of autoantibodies. Because on average 2% of transfusion recipients have an HPA-1a incompatibility and PTP is very rare, other factors probably influence the development of this syndrome. Thrombocytopenia is usually severe (platelet count <10,000/µL), and hemorrhage is common. A high-titer alloantibody is often detectable. Patients generally recover in a few weeks. However, life-threatening bleeding, including ICH, may occur. Prednisone and plasma exchange have been used, but IVIG is probably the treatment of choice for severe occurrences. Random-donor platelet infusions are ineffective and should not be administered. The efficacy of compatible, alloantigen-negative platelet transfusions is controversial.
Severe, transient thrombocytopenia has been associated with passive transfusion of plasma-containing products that have high-titer alloantibodies against HPA-1a or HPA-5b. This condition has been reported in neonates. The thrombocytopenia occurs within hours of transfusion. Most patients recover spontaneously, but treatment with IVIG or a glucocorticoid should be considered for severely affected patients.
Drug-Induced Immune Thrombocyopenia.
Numerous drugs have been implicated as causes of antibody-mediated thrombocytopenia. In general, the risk associated with any drug is low, but some drugs cause immune thrombocytopenia more commonly than others do. The thrombocytopenia generally resolves when use of the drug is discontinued, but the severity of bleeding symptoms and the duration of thrombocytopenia vary widely among patients. In some instances the sensitizing drug induces a second, drug-independent antibody (autoantibody) in addition to a drug-dependent one. Usually, these drug-induced autoantibodies are transient and disappear when treatment with the drug is discontinued. Occasionally, however, autoantibody production may be sustained and lead to a clinical picture indistinguishable from that of ITP. Once established, sensitivity to a drug generally persists indefinitely.
Pathogenesis.
Several mechanisms have been implicated in drug-induced immune thrombocytopenia.
Hapten Formation.
Small compounds can become covalently linked to blood cell membrane proteins and stimulate the production of antibodies specific for the hapten-protein complex. This mechanism is responsible for the hemolytic anemia seen in some patients treated with penicillin (see Chapter 13 ). Similarly, antibodies directed against hapten-protein complexes appear to be responsible for cases of penicillin-induced thrombocytopenia.
Compound Epitope Formation.
In patients with quinine-induced thrombocytopenia, antibodies that bind to normal platelets can be detected only when quinine is present in the soluble phase. These antibodies are usually specific for GPIIb/IIIa or GPIb/IX. The mechanism by which quinine promotes binding of antibody to platelets is not well understood. It is postulated that this agent binds noncovalently to one or more platelet membrane proteins and creates a “compound epitope” recognized by an antibody. Alternatively, the drug may interact with the target protein and cause a conformational change elsewhere in the molecule for which the antibody is specific. In addition to quinine, sulfonamide antibiotics and certain other drugs have been shown to induce antibodies that bind to platelets only when the sensitizing drug or one of its metabolites is present in the soluble state. Among children sulfamethoxazole is one of the more common causes of drug-induced immune thrombocytopenia.
Drug-Induced Autoantibodies.
Certain drugs, such as gold salts, procainamide, levodopa, and interferon alfa, induce true autoantibodies that bind to platelet membrane targets in the absence of the drug. The mechanisms by which these drugs induce autoantibodies is poorly understood.
Clinical Features.
Severe thrombocytopenia generally develops within 1 day of ingestion of quinine in a previously sensitized individual. A minimum of 1 week is required for initiation of the primary immune response in individuals taking this drug for the first time. Prodromal symptoms such as fever and chills may be observed. Bleeding may be severe, and its onset is abrupt. Hemorrhagic vesicles are common in the oral mucosa. The presence of a platelet antibody may be verified by a variety of techniques. Readministration of a suspected drug in an attempt to confirm an etiologic relationship is not recommended as a routine diagnostic measure.
Treatment.
Discontinuation of treatment with the offending drug is the main therapy. With quinine the thrombocytopenia usually begins to abate in 1 day if no additional drug is taken, and a normal platelet count is restored within 1 week. In patients with severe drug-induced thrombocytopenia, IVIG or a glucocorticoid has been used to ameliorate bleeding. However, there is little evidence that these medications are effective in this clinical setting. For life-threatening hemorrhage, platelet transfusion may be used, although this may be of limited value because of the persistence of slowly dissociating drug-antibody complexes. Because a sensitized individual is presumed to remain at lifelong risk for additional episodes of drug-induced thrombocytopenia, further use of the offending drug is contraindicated.
Heparin-Induced Thrombocytopenia and Thrombosis.
HIT is a complex clinical syndrome with an overall estimated incidence of 1% to 5% in adult patients exposed to heparin; the incidence in children is probably less, and it is virtually nonexistent in neonates. The thrombocytopenia usually begins 5 to 10 days after heparin therapy is started, but it can occur within hours if the patient previously received heparin. The risk of HIT is higher for therapeutic than for low drug doses, for bovine than for porcine heparin, and for unfractionated than for low-molecular-weight heparin (LMWH). In contrast to drug-induced thrombocytopenia caused by quinine or sulfonamides, severe thrombocytopenia (platelet count <10,000/µL) is rare in HIT. Typically, platelet counts range from 20,000 to 150,000/µL, with a median of 50,000/µL. The thrombocytopenia itself is rarely severe enough to cause bleeding symptoms and resolves after heparin therapy is discontinued. However, as many as 30% of patients with HIT experience arterial or venous thromboses. The development of thrombosis in patients with HIT is known as thrombocytopenia with thrombosis syndrome (HITTS), and this condition is associated with significant morbidity and mortality.
Pathogenesis.
The antibodies responsible for HIT recognize the complex of heparin and platelet factor 4 (PF4), an alpha granule constituent. After treatment with heparin, PF4 is released into the circulation and binds with heparin to form a reactive antigen on the platelet surface. The heparin-PF4 complex is immunogenic in some patients. IgG against this complex triggers platelet activation by binding to FcγRIIA receptors on the surface membrane. The activated platelets release substances that stimulate other platelets and result in thrombin generation. PF4 bound to glycosaminoglycans on the surface of endothelial cells may also serve as a target for antibody binding and lead to antibody-mediated injury to the endothelium and a predisposition to thrombosis.
Approximately 50% of patients exposed to heparin during cardiac catheterization and subsequently during cardiac surgery form antibodies against heparin-PF4 complexes, but HITTS develops in only a small percentage. It is unclear why heparin-PF4 complexes, which are composed of two substances normally present in the body, are so immunogenic. Moreover, it is not known why thrombocytopenia or thrombosis develops in some patients and not in others.
Diagnosis.
The diagnosis of HIT is based on the interpretation of clinical findings together with laboratory confirmation of heparin-PF4 antibodies. A scoring system has been proposed to assess the pretest probability of HIT, although this system has not been validated for pediatric patients. The gold standard laboratory test for diagnosing HIT is a functional test, the platelet [ 14 C]serotonin release assay. Unfortunately, the complexity and availability of this assay limit its usefulness. An enzyme-linked immunosorbent assay (ELISA) for detection of antibodies against heparin-PF4 complexes is available, and results obtained with the serotonin release assay and ELISA are generally in agreement in patients with a clinical diagnosis of HIT. However, ELISA has a more limited specificity and therefore produces false-positive results. A negative ELISA result generally excludes HIT. If the ELISA result is positive, a serotonin release assay may be performed to confirm the diagnosis or to invalidate a false-positive ELISA result.
Treatment.
It is standard practice to discontinue heparin in patients in whom HIT is suspected, but many of these patients will have thrombotic complications over the subsequent days. Therefore ongoing antithrombotic therapy is indicated. Patients with HIT should not be given warfarin as a substitute for heparin during the acute episode because this can cause an abrupt decrease in the level of protein C and put the patient at greater risk for thromboembolic complications such as warfarin-induced limb gangrene. LMWH is not recommended for the treatment of HIT because unfractionated heparin and LMWH preparations display significant platelet aggregation cross-reactivity in vitro and in vivo. Hirudin, a compound originally purified from the leech salivary gland, inactivates thrombin by forming a 1 : 1 complex. A recombinant hirudin, lepirudin, is effective in the treatment of HIT or HITTS. In children lepirudin should be started as a continuous infusion at 0.1 mg/kg/h without a preceding bolus, with a target activated partial thromboplastin time (APTT) of 1.5 to 2.5 times the patient’s baseline value (first assessed 4 hours after infusion). Other agents that are effective in this clinical setting are the synthetic antithrombin argatroban and the synthetic pentasaccharide fondaparinux. Danaparoid, a low-molecular-weight heparinoid consisting of a mixture of heparan sulfate, dermatan sulfate, and small amounts of chondroitin sulfate, has been used for more than a decade to treat patients with HIT. Danaparoid was voluntarily withdrawn from the United States market with the introduction of fondaparinux but is still available in other countries.
Thrombotic Microangiopathic Disorders.
The thrombotic microangiopathic spectrum of disorders is characterized by destructive thrombocytopenia, disseminated capillary thrombosis, and microangiopathic hemolytic anemia ( Box 34-4 ). Often organ dysfunction occurs because of microvascular thrombosis and ischemic necrosis. Thrombotic microangiopathic disorders may be primary or secondary. There are both acute and chronic (familial) relapsing forms.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


