Chapter 6 Acquired Disorders of Red Cell, White Cell, and Platelet Production

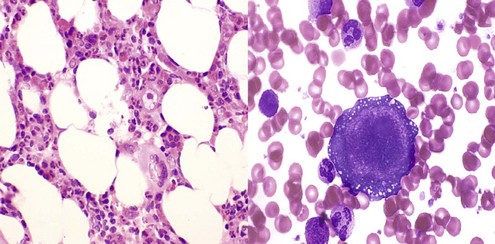
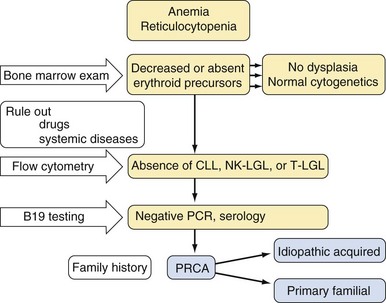
Table 6-2 Classification of Neutropenia
| Congenital | |
| Primary | Autoimmune neutropenia |
| Pure white cell aplasia | |
| Idiopathic | |
| Thymoma | |
| Hematologic malignancies (e.g., T-LGL leukemia) | |
| Infections/postinfectious | |
| Viral | |
| Measles,53 mumps, roseola,54,55 rubella,56 RSV, influenza57 | |
| Hepatitis A,58 B,58,59 and C60 | |
| CMV,61–63 EBV,64–66 HIV67,68 | |
| Parvovirus69–71 | |
| Bacterial | |
| Tuberculosis72,73 | |
| Brucellosis74–76 | |
| Tularemia77 | |
| Typhoid fever78 | |
| Rickettsial | |
| Rocky Mountain spotted fever79 | |
| Ehrlichiosis80,81 | |
| Fungal | |
| Histoplasmosis82,83 | |
| Parasitic | |
| Malaria,84 leishmaniasis85,86 | |
| Autoimmune conditions (e.g., SLE87,88, RA89) | |
| Drugs and chemicals | |
| Neutropenia associated with immunodeficiency90,91 | |
| Severe nutritional deficiencies92,93 | |
| Neutropenia due to increased margination | |
| Iatrogenic (e.g., hemodialysis94,95) |
CMV, Cytomegalovirus; EBV, Epstein-Barr virus; HIV, human immunodeficiency virus; RA, refractory anemia; RSV, respiratory syncytial virus; SLE, systemic lupus erythematosus; T-LGL, T-cell large granular lymphocyte.
Table 6-3 Drugs Associated With Agranulocytosis

ACE, Angiotensin-converting enzyme; IAAAS, International Agranulocytosis and Aplastic Anemia Study.
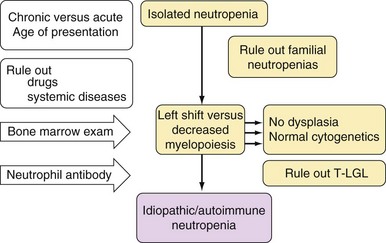
Table 6-4 Immunophenotype and Laboratory Features of T-Cell Large Granular Lymphocyte Leukemia
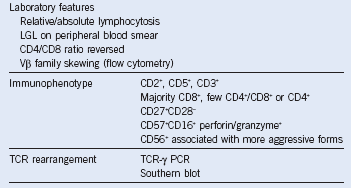
LGL, Large granular lymphocyte; PCR, polymerase chain reaction; TCR, T-cell receptor.
Approach to the Diagnosis and Treatment of Acquired Pure Red Cell Aplasia, Acquired Neutropenia, and T-Cell Large Granular Lymphocyte Leukemia
Related posts:
 Dynamic Interactions between Hematopoietic Stem and Progenitor Cells and the Bone Marrow: Current Biology of Stem Cell Homing and Mobilization
Clinical Manifestations and Treatment of Acute Lymphoblastic Leukemia in Children
Approach to Anemia in the Adult and Child
Infectious Mononucleosis and Other Epstein-Barr Virus–Associated Diseases: Part 2
Transfusion Therapy for Coagulation Factor Deficiencies
Diseases of Platelet Number: Immune Thrombocytopenia, Neonatal Alloimmune Thrombocytopenia, and Posttransfusion Purpura
Dynamic Interactions between Hematopoietic Stem and Progenitor Cells and the Bone Marrow: Current Biology of Stem Cell Homing and Mobilization
Clinical Manifestations and Treatment of Acute Lymphoblastic Leukemia in Children
Approach to Anemia in the Adult and Child
Infectious Mononucleosis and Other Epstein-Barr Virus–Associated Diseases: Part 2
Transfusion Therapy for Coagulation Factor Deficiencies
Diseases of Platelet Number: Immune Thrombocytopenia, Neonatal Alloimmune Thrombocytopenia, and Posttransfusion Purpura
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


