Acquired Hemorrhagic Disorders
Chapter 29 describes how the application of clinical skills forms the cornerstone for the assessment of a potential bleeding disorder. After confirming that a bleeding disorder is present through a comprehensive medical history and physical examination, the clinician should next address whether the condition is likely familial or acquired. This is sometimes difficult because some patients with mild underlying heritable hemostatic defects (e.g., mild von Willebrand disease [VWD] or platelet function defects) may not demonstrate suspicious clinical symptoms until faced with a traumatic injury or a surgical challenge at a later age. Nevertheless, acquired bleeding disorders remain a frequent clinical scenario, with presentations ranging from acute unexpected bleeding during or immediately after surgery to unusual or excessive clinical manifestations of bruising, petechiae, epistaxis, gum bleeding, or hematoma formation that occur over weeks to months. Distinguishing whether the bleeding most likely represents an abnormality in primary hemostasis, fibrin formation, or fibrinolysis provides a framework for the generation of a differential diagnosis (See Chapter 29 ). For example, petechiae are almost exclusively seen with defects in platelet number or function, whereas deep tissue hematomas are more likely associated with defects in fibrin formation, as would occur with clotting factor deficiencies. Standard screening laboratory tests such as the prothrombin time (PT) and activated partial thromboplastin time (aPTT) are most often used in the initial evaluation but are insensitive to mild but clinically significant reductions in hemostatic capacity. In addition, a platelet count does not provide any assessment of platelet function. Therefore the clinician must rely on the history and physical findings to guide the extent of additional coagulation studies. The most common causes of acquired hemorrhagic disorders include drug-induced bleeding (e.g., anticoagulants); disseminated intravascular coagulation; liver disease; vitamin K (VK) deficiency; massive transfusion; renal disease; and, rarely, acquired inhibitors to coagulation proteins.
Drug-Induced Bleeding
Many drugs have been associated with abnormal platelet function, although not all result in bleeding manifestations. The major classes include antiinflammatory agents, antibiotics, cardiovascular drugs, psychotropic drugs, anticonvulsants, and anticoagulants. Aspirin is the commonest example, acting by irreversibly acetylating a serine residue at the active site of cyclooxygenase, whereas other nonsteroidal antiinflammatories (e.g., ibuprofen, naproxen) are reversible inhibitors. β-Lactam antibiotics can interfere with platelet function through binding to the platelet membrane. Calcium channel blockers (e.g., nifedipine) and tricyclic antidepressants, such as amitriptyline, can result in decreased platelet aggregation responses but are unlikely to result in clinical bleeding. Valproic acid has become one of the most commonly used anticonvulsants in children. Interestingly, several mechanisms of relevant valproate-induced coagulopathies have been described, including thrombocytopenia and platelet dysfunction, acquired VWD, decreased VK-dependent clotting factors, hypofibrinogenemia, and decreased factor XIII levels. Therapeutic anticoagulants (heparin, low-molecular-weight heparin, warfarin, and direct inhibitors of factor Xa and thrombin) should be the most readily apparent cause of drug-induced bleeding and in most patients can be correlated with laboratory monitoring. Platelet receptor antagonists (IIb/IIIa inhibitors; e.g., abciximab, eptifibatide, tirofiban, and adenosine diphosphate receptor antagonists; e.g., clopidogrel) are in increasing use in patients with congenital heart disease and patients undergoing interventional studies.
In most cases discontinuation of the offending drug should resolve the hemorrhagic manifestations, but this will depend on the reversibility and half-life of the drug. VK administration will in most cases rapidly reverse warfarin-induced bleeding. Alternatively, when more rapid replacement is necessary to correct low levels of factors II, VII, IX, and X, infusion with a prothrombin complex concentrate (PCC) or fresh frozen plasma (FFP) can be considered. PCCs are formulated with either three factors (II, IX, and X) or four factors (II, VII, IX, and X) and have some advantages over FFP. PCCs can be rapidly reconstituted in small infusion volumes, have a fast onset of action, do not require identification of the patient’s blood group, have an exceptional safety profile with respect to risk of viral transmission owing to pathogen reduction and inactivation steps incorporated into their manufacturing, and reduced risk of adverse reactions such as transfusion-associated circulatory overload or transfusion-associated acute lung injury. A four-factor PCC, Kcentra, was recently approved by the U.S. Food and Drug Administration (FDA) for the urgent reversal of acquired coagulation factor deficiency induced by VK antagonism therapy in adults with acute major bleeding. Transfusions of fresh platelets are indicated in those patients with life-threatening or unremitting bleeding caused by drug-induced platelet dysfunction. In cases of minor bleeding in which the drug cannot be readily discontinued (e.g. anticonvulsants), desmopressin has been used with some success.
Disseminated Intravascular Coagulation
Disseminated intravascular coagulation (DIC) is a pathologic syndrome in which the normal physiology of coagulation is disturbed by the simultaneous action of four mechanisms: increased thrombin generation, suppressed physiologic anticoagulant pathways, activation and subsequent impairment of fibrinolysis, and activation of the inflammatory pathway. This leads to widespread intravascular deposition of fibrin with resultant thrombotic end-organ complications and consumption of platelets and coagulation proteins, resulting in severe bleeding. Associated damage to the microvasculature can contribute to organ dysfunction, capillary leak, and shock.
Etiology
DIC is always a secondary phenomenon and not a disease entity in its own right. Thus its recognition should prompt the clinician to identify and treat the underlying cause rather than merely react to the bleeding manifestations. It most frequently occurs in the settings of sepsis, trauma, and systemic inflammatory syndrome, with an approximate frequency in hospitalized children of 0.4% to 1%, with sepsis accounting for approximately 95% of cases. It is primarily a clinical diagnosis based on the evaluation of laboratory results in patients with a clinical condition known to be associated with DIC ( Box 35-1 ).
Infectious
Meningococcemia (purpura fulminans)
Other gram-negative bacteria (Haemophilus, Salmonella)
Gram-positive bacteria (group B Streptococcus)
Rickettsia (Rocky Mountain spotted fever)
Viruses
Malaria
Fungus
Tissue Injury
Central nervous system trauma (massive head injury)
Multiple fractures with fat emboli
Crush injury
Profound shock or asphyxia
Hypothermia or hyperthermia
Massive burns
Malignancy
Acute promyelocytic leukemia
Acute monoblastic or myelocytic leukemia
Widespread malignancies (neuroblastoma)
Venom or Toxin
Snake bites
Insect bites
Microangiopathic Disorders
“Severe” thrombotic thrombocytopenic purpura
Hemolytic-uremic syndrome
Giant hemangioma (Kasabach-Merritt syndrome)
Gastrointestinal Disorders
Fulminant hepatitis
Severe inflammatory bowel disease
Reye syndrome
Hereditary Thrombotic Disorders
Homozygous protein C deficiency
Miscellaneous
Severe graft rejection
Acute hemolytic transfusion reaction
Severe collagen-vascular disease
Kawasaki disease
Heparin-induced thrombosis
Infusion of “activated” prothrombin complex concentrates
Hyperpyrexia/encephalopathy, hemorrhagic shock syndrome
Clinical Presentation
The clinical manifestations of DIC include bleeding, thrombosis, or both, however typically bleeding predominates. Early indicators are bleeding resulting from venipunctures, intravascular accesses, and surgical wounds. Mucocutaneous bleeding may manifest as bruising, petechiae, epistaxis, gum bleeding, blood from tracheal aspirates, gastrointestinal bleeding, and hematuria. In fulminant cases it may lead to bleeding into vital organs. However, with increasingly sensitive diagnostic tests that can detect endogenous activation of the hemostatic process, the clinical spectrum of DIC is broad. Nonovert DIC describes a stressed but compensated hemostatic system, in which the laboratory tests are abnormal but there are no clinical manifestations. Overt DIC is described as a stressed and decompensated hemostatic system, in which the laboratory tests are abnormal and clinical bleeding or microvascular thrombosis and organ dysfunction exists. Overt DIC may be controlled or uncontrolled depending on whether the process will resolve when the underlying stimulus is removed.
Diagnosis
The clinical presentation of a profoundly ill child with bleeding believed to be secondary to DIC can be supported by laboratory tests showing evidence of a consumptive coagulopathy with activation of the fibrinolytic cascade. Moderate to severe thrombocytopenia with or without anemia will be evident from the complete blood count. Thrombocytopenia is present in approximately 50% of patients and suggests consumption of platelets. Anemia could be caused by bleeding or, when accompanied by schistocytes on blood smear, evidence of microangiopathic hemolytic anemia. The PT and aPTT are prolonged in 50% to 60% of patients, reflecting consumption of many coagulation proteins, including prothrombin; factors V, VII, and VIII; and fibrinogen. Fibrinogen or fibrin degradation products (FDPs) and D-dimers are both increased in concentration in most patients with DIC, suggesting activation of the fibrinolytic process. Marked reductions in plasma anticoagulants including proteins C and S and antithrombin (AT) have also been described. The most sensitive tests for diagnosis of DIC are markers of endogenous thrombin generation: prothrombin fragment 1.2 and thrombin-AT (TAT) complexes. Prothrombin fragment 1.2 is released when thrombin is generated from prothrombin. TAT complexes are generated by binding of thrombin with its inhibitor AT. The standard assays (PT, aPTT, platelet count, and D-dimers) are relatively rapid and simple to perform. However, changes in these test results do not always occur at the same time, and laboratory values change rapidly depending on the patient’s clinical status. This may create confusion in patient management and make the diagnosis of DIC at an early stage particularly difficult. The International Society on Thrombosis and Haemostasis (ISTH) diagnostic scoring system for overt DIC has been widely used in intensive care units, and several publications have validated the score against morbidity scores. The five-step algorithm assigns a score based on the severity of abnormality for each of the following: platelet count (>100 × 109/L = 0; <100 × 109/L = 1; <50 × 109/L = 2), elevated fibrin-related markers (no increase = 0; moderate increase = 2; strong increase = 3), prolonged prothrombin time (<3 seconds = 0; <3 seconds but <6 seconds = 1; >6 seconds = 2), and fibrinogen level (>1 g/L =0; <1 g/L = 1). A total score of 5 or more is considered compatible with overt DIC. The sensitivity and specificity of this scoring system are greater than 90%. However, the algorithm should be applied only in the presence of an underlying disorder known to be associated with DIC. Despite its use in many pediatric intensive care units in the United States, there remains a paucity of clinical research studies examing the utility of the ISTH scoring system in children. Investigators at Texas Children’s Hospital have compared their institutional modified DIC criteria with the ISTH diagnostic score against a gold-standard diagnostic method of confirmation of DIC in a subset of children who died. Their modified criteria is not a scoring system but a sequential analysis of coagulation assays (PT, platelet count, fibrinogen, and D-dimer) as evaluated by transfusion medicine specialists along with the patient’s clinical condition. Such an approach yielded a higher sensitivity, owing to the inclusion of sequential testing, to recognize a trend in the evolution from an early-phase DIC to overt DIC.
Treatment
The fundamental principal of DIC treatment is the specific and vigorous treatment of the underlying disorder. In some cases DIC will completely resolve within hours after resolution of the underlying condition (e.g., appropriate control of sepsis with antimicrobials). However, in other cases supportive measures are required to control the DIC until the underlying condition is resolved (e.g., the use of all- trans -retinoic acid and chemotherapy for the treatment of acute promyelocytic leukemia and DIC. ) Therapeutic interventions remain controversial and must be individualized according to the underlying basis for the DIC and severity of the clinical symptoms. For example, in nonovert DIC, children do not usually require therapy for the DIC itself. However, in the presence of uncontrolled overt DIC, therapeutic intervention including blood replacement products may be indicated to improve hemostasis while waiting for effective therapy for the underlying condition. Treatment modalities investigated include blood component therapy, anticoagulants, restoration of natural anticoagulant pathways, and other agents.
Blood Component Therapy.
In general, the more severe the laboratory abnormalities, in particular the degree of thrombocytopenia and coagulation factor depletion, the greater the risk of bleeding complications with DIC. Hence treatment with FFP, fibrinogen, cryoprecipitate, and platelets appears to be a rational therapy in bleeding patients or patients who are at risk for bleeding with a significant depletion of these hemostatic factors. However, blood component therapy should not be instituted on the basis of laboratory results alone; it is indicated only in patients with active bleeding, those who require an invasive procedure, or those who are otherwise at risk for bleeding complications. Large volumes of plasma (15 to 30 mL/kg) may be necessary to correct the coagulation defect. PCC may be considered in actively bleeding patients if FFP transfusion is not possible. However, these products lack certain essential coagulation factors, such as factor V. The efficacy and safety of recombinant factor VIIa in DIC with life-threatening bleeding are unknown. Reasonable goals are to maintain platelet counts above 50 × 10 9 /L, fibrinogen concentrations above 1.5 g/L, and PT values less than double the normal range.
Anticoagulant Therapy.
Considering the central role played by thrombin in DIC, the use of heparin or other anticoagulants to inhibit thrombin generation appears reasonable. Heparin can at least partly inhibit the activation of coagulation in sepsis and other causes of DIC. However, a beneficial effect of heparin on clinically important outcome events in patients with DIC has never been demonstrated in controlled clinical trials and is not a standard of care in overt cases of DIC. However, therapeutic doses of heparin may be indicated in patients with clinically overt thromboembolism, chronic DIC, or extensive fibrin deposition such as seen in purpura fulminans or acral ischemia.
Replacement of Natural Anticoagulant.
Depleted levels of AT, protein C, and protein S cannot be effectively replaced with FFP alone because of the short plasma half-life of these proteins. AT and protein C concentrates have been extensively evaluated in patients with DIC. Trials of AT therapy have not yet provided conclusive evidence sufficient to make treatment recommendations. The double-blind, placebo-controlled, phase III trial of recombinant human activated Protein C Worldwide Evaluation in Severe Sepsis (PROWESS) demonstrated a significant decrease in mortality for the protein C–treated group compared with placebo. However, these findings were not replicated in later trials, treatment was associated with a higher risk of bleeding, and recombinant human protein C was ultimately withdrawn in 2011. Soluble recombinant human thrombomodulin acts by reducing thrombin-mediated clotting, enhancing protein C activation at the site of clotting, as well as demonstrating some antiinflammatory properties. However, a large randomized controlled trial failed to demonstrate a benefit in 28-day mortality rates.
Other Agents.
Recently, recombinant factor VIIa (rVIIa) has become an attractive strategy to control bleeding in various scenarios. In situations where volume overload is an issue or bleeding persists despite adequate blood component support, use of rVIIa has been shown to be effective. However, these data have mostly been generated from anecdotal reports, and given the potential adverse thrombotic complications with this agent, controlled randomized trials are required to address its safety and efficacy in these patients. Antifibrinolytic agents can be effective in bleeding patients, but the use of these agents in patients with bleeding associated with DIC is generally not recommended. They could be considered in cases when hyperfibrinolysis is suspected, such as may occur with acute promyelocytic leukemia or trauma.
Liver Disease
Pathophysiology
The liver is the main site of synthesis for most hemostatic components. However, the hemostatic impairment in liver disease involves a variety of mechanisms, including impaired hepatic synthesis, activation of the coagulation and fibrinolytic systems, poor clearance of activated hemostatic components, loss of hemostatic proteins into ascitic fluid, concurrent VK deficiency, thrombocytopenia, and impaired platelet function. When both procoagulants and anticoagulants are reduced, hemostatic dysregulation results in both a propensity to hemorrhage and a propensity to thrombose. Along with reduced synthesis there may be secretion of abnormal forms of the hemostatic proteins that may function as inhibitors to coagulation. For example, abnormal fibrinogens (dysfibrinogenemias) are very common in liver disease. Similarly, VK-dependent proteins (factors II, VII, IX, X, protein C, protein S, and protein Z) decrease in liver disease. However, they may also be secreted in forms with abnormal gamma-carboxylation reactions of the glutamic acid residues on the amino-terminal portion of the protein leading to reduced functional activity. Hypofibrinogenemia may be related to decreased synthesis, or increased consumption (e.g., DIC). However, in patients with acute liver failure, plasminogen activator inhibitor (PAI-1) is increased, shifting the balance toward hypofibrinolysis. This occurs despite elevated levels of tissue plasminogen activator (tPA) that may be secondary to increased release from the activated endothelium or reduced clearance by the diseased liver (or both). The other proteins involved in fibrinolysis (plasminogen, α2-antiplasmin, thrombin activatable fibrinolysis inhibitor, and factor XIII) are typically reduced because of decreased liver synthesis. In addition, several structural manifestations of liver disease can contribute to the bleeding in these patients, including portal hypertension, varices, gastritis, and hemorrhoids.
Clinical Presentation
Clinical symptoms are variable and dependent, to some extent, on the etiology of the liver failure and associated invasive procedures. Symptoms include ecchymosis and petechiae, mucous membrane bleeding, hemorrhage from gastrointestinal varices, and hemorrhage into the abdomen or central nervous system (CNS). However, clinically significant bleeding occurs rarely in acute liver failure (approximately 5% to 10% of cases). Coagulation screening tests (aPTT, PT, thrombin clotting time) are usually prolonged, platelet counts are reduced, and bleeding time is prolonged. Plasma concentrations of FVII; FV; and, less commonly, fibrinogen are decreased. Levels of FVIII may be normal or elevated, possibly reflecting reduced clearance via low-density-lipoprotein receptor-related protein, and can be helpful in distinguishing severe liver disease from DIC. However, both conditions may occur simultaneously. FDPs or D-dimer levels are often increased in hepatic failure and contribute to prolongation of the thrombin clotting time and impaired platelet function. Dysfibrinogenemia will contribute to prolongations of the PT and aPTT, but the thrombin time and reptilase assays are more sensitive. Dysfibrinogenemia may be confirmed by demonstrating an abnormal ratio of clottable fibrinogen (functional assay) to fibrinogen antigen (immunoassay).
Treatment
Treatment for the hemostatic manifestations of severe hepatic failure is difficult. Enhanced hemostasis after replacement therapy is usually transient because adequate volumes of FFP cannot be infused, owing to associated hypervolemia. In addition, many coagulation proteins are rapidly cleared, thereby limiting any beneficial effect. Usually, therapy specifically directed at the coagulopathy is reserved for active bleeding or to facilitate invasive procedures. A common indication for FFP is an international normalized ratio (INR) greater than 2 or a PT prolongation greater than 4 seconds. However, there are no firm guidelines, and some interventions may safely be performed with INR in the 2 to 2.5 range. Although desmopressin can induce the release of factor VIII and von Willebrand factor (VWF) with a shortening of the bleeding time, in randomized trials desmopressin failed to control bleeding from acute variceal hemorrhage or decrease blood loss with hepatectomy. Cryoprecipitate can be administered to increase plasma fibrinogen concentrations if necessary. Secondary VK deficiency owing to impaired VK utilization or impaired VK absorption owing to cholestasis from intrahepatic and extrahepatic biliary atresia requires treatment with VK. Isovolumetric plasmapheresis may aid in restoration of hemostasis by replacing coagulation factors while removing activated clotting factors, tPA, fibrin, and FDPs. Recombinant VIIa has been shown to correct the prolonged PT in nonbleeding cirrhotic patients and has been used to avoid volume overload before invasive procedures likely to cause bleeding. However, randomized trials in the setting of liver biopsy and variceal bleeding have shown either no difference in hemostasis or only early, modest reductions in rebleeding rate but no difference in overall bleeding or transfusion requirements. Larger studies are still required to evaluate the efficacy and safety of this agent in liver disease.
Patients requiring liver transplantation invariably have severe, end-stage hepatic failure with its associated hemorrhagic complications. Additional risk factors that contribute to bleeding in children during transplantation include extensive scar tissue from previous surgery, prolonged duration of the surgery, the need to reduce the size of the donor organ to fit the small patient, dilutional coagulopathy, and excessive activation of the fibrinolytic system during the anhepatic phase. Although treatment with antifibrinolytic agents is an attractive option, this approach should be used with caution because children who undergo liver transplant may later develop either hemorrhagic or thrombotic complications. The prophylactic use of recombinant VIIa to reduce blood loss and transfusion requirements in patients undergoing liver transplantation has been studied in two multicenter randomized controlled trials. These trials failed to demonstrate a benefit to support its use as a universal preemptive therapy for liver transplantation. Recombinant VIIa could be considered in individual cases as “rescue therapy” for patients with life-threatening coagulopathy and bleeding during transplantation or in situations where blood products are unavailable or unacceptable (e.g., the Jehovah witnesses).
Vitamin K Deficiency
The role of VK in coagulation is detailed in Chapter 27 , and VK deficiency bleeding in newborns is discussed in detail in Chapter 29 .
Clinical Presentation
Clinical symptoms of VK deficiency include mild to moderate bleeding; ecchymoses; oozing from intravenous (IV) puncture sites; and, rarely, internal bleeding. The primary disorder is usually identified on the basis of the history and physical examination.
Primary VK deficiency in healthy children is uncommon because of a relatively low VK requirement and the widespread distribution of VK in plant and animal tissues. In plants the only important molecular form is phylloquinone (VK1). Bacteria synthesize a family of vitamin K compounds called menaquinones (VK2). The contribution of the microbiologic flora of the gut to VK dietary intake is unknown but may be as high as 50%. Although broad-spectrum antibiotics may reduce VK2 production by intestinal bacteria, VK deficiency is rare in the presence of a normal diet.
Groups of children at high risk for VK deficiency include breastfed newborns, chronically ill children with an inadequate dietary intake of VK, children with disorders that interfere with the absorption of VK (e.g., diarrhea, cystic fibrosis, cholestatic liver disease, celiac disease), and children with poor nutrition who are receiving broad-spectrum antibiotics. Cystic fibrosis, biliary atresia, and obstructive jaundice interfere with the absorption of fat-soluble vitamins and interfere with transport to the liver, with resultant VK deficiency. Therapeutic drugs can interfere with VK metabolism (e.g., phenytoin, warfarin). However, at least in the case of warfarin administration, this is an expected side effect. Accidental exposure to supratherapeutic doses may occur in small children with access to warfarin in the home. Anticoagulant rodenticides can be another source of accidental exposure. Owing to the emergence of warfarin-resistant strains of rats, newer “long-acting” second-generation compounds have been utilized. These so-called “superwarfarins” have higher affinity for vitamin K1-2,3 epoxide reductase, are able to disrupt the vitamin K1-epoxide cycle at more than one point, exhibit hepatic accumulation, and have unusually long biological half-lives as a result of their high lipid solubility and enterohepatic circulation. After accidental ingestion these compounds can cause anticoagulation for several days or even weeks. However, in most cases involving small children, the exposure is low and bleeding manifestations are rare. VK deficiency has been described in bone marrow transplant and oncology patients in whom the etiology is multifactorial as a result of drug antagonism, liver dysfunction, fat malabsorption, anorexia, or inadequate dietary intake. A study of children evaluated before undergoing bone marrow transplantation identified 31% (8 of 26) with evidence of VK deficiency (as evidenced by circulating PIVKA-II), although only one patient had a prolonged PT, highlighting that the PT alone is an insensitive marker of VK deficiency. Of the eight affected patients, six exhibited bleeding manifestations peri-transplant.
Laboratory Diagnosis
The PT and aPTT may both be abnormal in VK deficiency but should correct in a 1 : 1 mix with normal plasma. Other acquired or congenital coagulopathies can give similar screening test abnormalities. Therefore specific coagulation factor assays must be performed. The finding of more than one VK-dependent factor deficiency should increase the suspicion of an acquired deficiency. A normal factor V in addition to normal liver function studies would suggest that there is no significant liver disease underlying the presentation. A normal FVIII and fibrinogen level and absence of elevated D-dimers would make DIC unlikely. However, VK deficiency can complicate other coagulopathies, such as DIC or liver disease. If there is any doubt about the diagnosis, the patient should receive VK therapy in conjunction with other supportive care.
Treatment
The route and specific type of therapy are dictated by the urgency of the clinical situation and potential side effects of therapy. For example, severe anaphylactoid reactions, although rare, have complicated IV VK administration, even when the solution is diluted and infused slowly. Therefore IV administration of VK, at a rate no faster than 1 mg per minute, should be restricted to those situations in which other routes are not feasible and the risk is justified. Intramuscular VK is not recommended because it can result in pain, swelling, and hematoma formation. The preferred systemic route is subcutaneous because it is safe and effective. Oral VK is effective if absorption is unimpaired. Although oral administration is generally thought to produce a slower correction of the PT (6 to 8 hours) compared with parenteral VK (2 to 6 hours), a study in adults suggested that the oral route may actually be faster, at least in reversing the effect of warfarin.
Asymptomatic patients with mildly abnormal coagulation results that are presumed secondary to VK deficiency should be given VK subcutaneously (1 to 5 mg depending on size). This approach is both therapeutic and diagnostic, with coagulation abnormality correction within 2 to 6 hours. A patient bleeding because of VK deficiency should receive 2 to 10 mg of VK subcutaneously. FFP (10 to 20 mL/kg) is particularly useful when the precise nature of the coagulopathy is unknown and bleeding is severe. However, the amount of plasma needed for total correction of a severe VK deficiency is so great that it may result in volume overload. A patient with a life-threatening hemorrhage or an intracranial hemorrhage (ICH) would likely benefit from prothrombin complex concentrates at a minimum dose of 50 units/kg, in addition to systemic VK (5 to 20 mg). Prothrombin complex concentrates contain relatively uniform amounts of factors II, IX, and X, with amounts of factor VII/VIIa varying. Recombinant VIIa has been used anecdotally.
Prevention
Prophylactic VK should be given to patients at risk, such as patients with inadequate nutrition and those who are receiving broad-spectrum antibiotics. Most patients requiring total parenteral nutrition (TPN) are supplemented with VK. There has been concern in the past that maternal anticonvulsant therapy may be associated with increased risk of neonatal bleeding linked to enzymatic degradation of VK. This has prompted recommendations for VK prophylaxis to pregnant mothers on anticonvulsants. However, recent studies have demonstrated that the incidence of bleeding in infants exposed to these drugs antenatally is not higher than controls and have shown no benefit to routine VK prophylaxis.
Massive Transfusion Coagulopathy
Uncontrolled bleeding can be a clinical problem associated with the management of trauma and surgical patients, often necessitating the transfusion of large amounts of blood and blood derivatives. Massive transfusion has been commonly defined as replacing at least one blood volume in 24 hours or 50% of one blood volume in 3 hours. Criteria used by the University of Michigan Hospitals for adults is 4 U of red blood cells utilized in less than 4 hours and ongoing uncontrolled bleeding, and for children 30 mL/kg of red blood cells utilized and ongoing uncontrolled bleeding. Patients with this degree of bleeding will develop defective hemostasis related to the transfusions in addition to the underlying precipitating trauma or surgical insult. The resulting bleeding diathesis may be complex and may include evidence of DIC; depletion of hemostatic factors through blood loss, tissue injury, and consumption of factors; dilutional coagulopathy owing to aggressive blood component resuscitation; hypothermia; platelet dysfunction; and excessive fibrinolysis. Hemostatic resuscitation advocates for the transfusion of red blood cells, plasma, and platelets in a 1 : 1 : 1 ratio in order to address the underlying dilutional coagulopathy. Massive transfusion protocols are now utilized worldwide for patients with severe life-threatening bleeding and are intended to standardize the approach to these patients and provide structure and organization to what is usually a chaotic situation. These protocols standardize the ratio for blood products that are empirically delivered; initiate unique communication processes among the blood bank, the clinical laboratories, and the clinician; and call attention to appropriate management of acidosis, hypothermia, and hypocalcemia that may be aggravating the coagulopathy. Clinical studies with massive transfusion protocols have demonstrated decreased crystalloid use, improved survival rates, decreased chance of organ failure, and decreased risk of early death resulting from hemorrhage. Fibrinogen concentrates for acquired fibrinogen deficiency have been evaluated by postmarketing surveillance studies and retrospective analyses and show results consistent with in vitro studies demonstrating that fibrinogen replacement can improve clot firmness and improve survival of severely injured massively bleeding patients. Recombinant factor VIIa has been evaluated in several randomized clinical trials for bleeding in the context of trauma and surgery but has failed to improve outcomes.
Acquired Inhibitors of Coagulation Proteins
Inhibitors directed against coagulation factors are circulating antibodies that specifically neutralize the procoagulant activity or increase the clearance of those coagulation factors leading to a plasma deficiency and are often accompanied by bleeding manifestations. They may be either alloantibodies or autoantibodies. Examples of alloantibodies include those directed against infused factor VIII and are observed in 15% to 25% of patients with hemophilia A (discussed in Chapter 31 ). Autoantibodies have also been observed directed against endogenous factor VIII in nonhemophiliacs and occur typically in postpartum women, elderly individuals, or people who have autoimmune disorders. However, autoantibodies have also been ascribed to other coagulation proteins, including fibrinogen; factors II, V, VII, IX, X, XI, and XIII; andVWF. Antiphospholipid antibodies (APLAs), or lupus anticoagulant (LA), are a heterogeneous group of antibodies that react with proteins bound to phospholipids. Although these primarily function as inhibitors to in vitro phospholipid-dependent clotting assays, they paradoxically predispose to thrombosis rather than hemorrhage. However, a unique LA coagulopathy has been observed with a hemorrhagic syndrome associated with hypoprothrombinemia (discussed later).
Acquired Inhibitors to Factor VIII
Anti–factor VIII autoantibodies are rare in children but can result in severe bleeding and significant morbidity. They are associated with underlying medical conditions in half of those affected, including malignancy, autoimmune disease, lymphoproliferative disorders, and drugs (e.g., penicillin). Patients can present with bleeding symptoms ranging from easy bruising to intracranial hemorrhage. In one patient the clinical course was also complicated by nephrotic syndrome. Laboratory screening studies will show a prolonged aPTT that does not fully correct on mixing with normal plasma in a 1 : 1 mix (“Inhibitor Screen”). In addition, this can be distinguished from an LA by performing the aPTT 1 : 1 mix again with an incubation phase (typically at least 30 minutes at 37° C) in which the aPTT will show further prolongation indicating progressive neutralization of factor VIII provided by the normal plasma mixing. There will be no significant additional prolongation seen on incubation in the presence of an LA. In contrast to congenital hemophilia with inhibitors, these antibodies typically demonstrate type II (nonlinear) inactivation kinetics. Thus the baseline factor VIII levels may be higher than 1% despite high titer inhibitory antibodies.
Treatment is two-pronged, directed at stopping the bleeding and eradicating the inhibitor. Hemostasis can be achieved with high doses of factor VIII concentrates, activated prothrombin complex concentrates (APCC), and recombinant VIIa. Immunosuppression may be accomplished with steroids alone or in combination with mycophenolate-mofetil, rituximab, γ-globulin, or cytotoxic agents such as cyclophosphamide. Inhibitors have also been eradicated through extracorporeal immunoadsorption and immune tolerance with regular exposure to high-dose factor VIII infusions. Review of the available literature suggests that, in contrast to adults, anti–factor VIII autoantibodies in children may resolve more quickly and easily and do not necessarily require steroids and cytotoxic agents. Recombinant factor VIII should be considered in the initial treatment with additional hemostatic support from APCCs and recombinant VIIa as needed. Withdrawal of instigating drugs such as penicillin may also facilitate resolution.
Acquired Inhibitors to Factor IX
Few cases of spontaneous factor IX inhibitors have been described in nonhemophiliac patients, and most adult cases have been related to underlying systemic disorders such as systemic lupus erythematosus, hepatitis, multiple sclerosis, rheumatic fever, collagen vascular disease, postpartum status, and postprostatectomy status. Reports in children are limited to a few case reports. Bleeding manifestations included cutaneous ecchymoses and soft tissue hematomas. Diagnostic studies are similar to those with anti–factor VIII inhibitors. Treatments have included corticosteroids, γ-globulin, and cyclophosphamide, with rapid resolution of the inhibitor. One child had a spontaneous resolution. Recombinant factor IX and recombinant VIIa can be considered for acute management of bleeding.
Acquired Inhibitors to Factor V
Most cases of inhibitors directed against factor V develop as a result of risk factors such as surgical procedures, antibiotic administration (typically the β-lactam group), blood transfusions, cancers, and autoimmune disorders. However, the majority of cases arise after exposure to bovine thrombin. Topical preparations of bovine thrombin are widely used for surgical hemostasis and contain fibrinogen as well as small amounts of factor V and other proteins. Acquired inhibitors to coagulation factors, as a consequence of exposure to this highly immunogenic agent, is a frequent occurrence, albeit often unrecognized. In this context antibodies directed against factor V are most frequent, although anti-prothrombin and anti–factor X antibodies have also been described. The incidence of symptomatic inhibitors is not known. In a study of 151 adult patients exposed to bovine thrombin during cardiac surgery, 56% of the patients developed antibodies directed against human coagulation proteins. However, adverse clinical outcomes did not correlate with antibody formation. A literature review identified 12 cases of inhibitors in pediatric patients after thrombin exposure. In eight cases the antibodies were directed against human coagulation proteins; five of these patients had hemorrhagic complications. Bleeding manifestations included cutaneous, gastrointestinal, pulmonary, and cerebral hemorrhages. Patients exhibited both PT and aPTT prolongations and mixing studies consistent with an inhibitor. Specific factor activity assays revealed the target for the inhibitor. However, there was no correlation between laboratory studies and clinical bleeding. Corticosteroids and γ-globulin have been used with resolution of the inhibitor over days to weeks. Platelet transfusions are another option, insofar as approximately 20% of factor V activity is present in platelet granules that are shielded from circulating antibodies. Coagulation factor inhibitors should be suspected in any postoperative patient exposed to topical bovine thrombin presenting with a prolonged PT and aPTT.
Acquired Inhibitors to Other Coagulation Factors
Antifibrinogen antibodies are extremely rare and may or may not be associated with clinical bleeding. An increased rate of fibrinogen antibodies has been described in pregnant and peripartum women. Acquired inhibitors directed against coagulation factors VII and X have been observed with malignancies, autoimmune disorders, and drug exposure. In addition, acquired inhibitors against factor XI have been rarely reported, including in patients with underlying SLE.
Acquired von Willebrand Syndrome
Acquired von Willebrand syndrome (AVWS) is a rare bleeding disorder with clinical and laboratory findings similar to those in inheritedVWD. Since its original description in 1968 in a patient with systemic lupus erythematosus, fewer than 700 cases have been reported. AVWS usually occurs in individuals with no personal or family history of VWD and is accompanied by bleeding symptoms in about three quarters of patients, whereas others are diagnosed on the basis of abnormalities found during routine hemostasis screening tests. As would be expected, mucocutaneous bleeding symptoms predominate. Underlying disorders are identified frequently and fall into the following main categories: lymphoproliferative and myeloproliferative disorders; solid tumors; immunologic and cardiovascular disorders; and other miscellaneous conditions, including drug associations. It is typically a syndrome of advanced age, although it has also been reported in children, and is primarily associated with congenital heart disease, collagen vascular diseases, Wilms tumor, hypothyroidism, and use of certain drugs. Drug-associated AVWS has been described in up to 20% of pediatric patients who received valproic acid, although bleeding manifestations were mild and did not require discontinuation of the anticonvulsant.
Four main pathogenic mechanisms can lead to AVWS: (1) reduced VWF synthesis (as seen in severe hypothyroidism or certain drugs (e.g., valproic acid); (2) inhibition or clearance by paraproteins or autoimmune inhibitors (this has been observed in association with B-cell lymphomas, monoclonal gammopathies, multiple myeloma, and autoimmune disorders); (3) absorption of VWF high-molecular-weight multimers onto malignant cell clones or platelets; (4) increased shear stress and proteolysis, as may occur with aortic stenosis, artificial heart valves, left ventricular assist devices, or other cardiac defects with disturbed flow. Laboratory studies rely on detecting abnormally reduced functional VWF studies (ristocetin cofactor activity, collagen-binding assay) in comparison to VWF antigen and a selective deficiency of the high-molecular-weight multimers. A search for evidence of an inhibitor should be performed by measuring VWF activity after mixing experiments involving patient plasma and normal plasma incubated at 37° C.
The three goals of treatment should be control of active bleeding, prevention of bleeding with necessary invasive procedures, and treatment of the underlying disease. Desmopressin and factor VIII/VWF concentrates have provided only short-term control of bleeding in the presence of autoantibodies. In those cases γ-globulin, corticosteroids, or other immunosuppressive agents have been administered. Recombinant VIIa may also be effective with this presentation. In noninhibitor-associated AVWS, factor VIII/VWF concentrates are effective in most patients, although the half-life may be shorter. Desmopressin may be used to control bleeding, although there is a relatively lower rate of success among patients with cardiovascular or myeloproliferative disorders. Ultimately, normalization of hemostasis may not be seen until the underlying precipitating condition is treated effectively (e.g., with surgery or chemotherapy for Wilms tumor).
Acquired Inhibitors to Prothrombin—Hemorrhagic Lupus Anticoagulant Syndrome
LA represent a diverse group of antibodies directed against proteins bound to phospholipids. They may be found in patients with systemic lupus erythematosus and other autoimmune disorders but also in otherwise healthy individuals and have been implicated in thrombotic complications. However, in children they are most often identified after investigation of a prolonged aPTT seen as part of pre-surgical evaluation or associated with a history of infection. In the vast majority of these cases, they are asymptomatic (including no bleeding manifestations), transient, usually resolving over days to weeks, and do not preclude proceeding with invasive procedures. However, a rare hemorrhagic LA syndrome (ecchymoses, epistaxis, gastrointestinal bleeding, hematomas and even hemarthroses) has been observed with associated low prothrombin levels. This would be manifest by a concurrent prolongation of the PT (unusual for an uncomplicated LA) and pathogenically is believed to be caused by antiprothrombin antibodies. These are not typically neutralizing and, as a result, mixing studies may not indicate the presence of an inhibitor. Rather, the formation of immune complexes leads to increased plasma clearance. Antibodies have been characterized directed against the carboxy terminal portion of the prothrombin molecule. Bleeding symptoms usually resolve without therapy, although patients with significant bleeding symptoms may be treated with prothrombin complex concentrates. Corticosteroids and other immune suppressants have been used in some cases.
Acquired Thromboembolic Disease
Thromboembolism (TE) in children has become increasingly diagnosed in recent years thanks to enhanced survival rates associated with serious underlying illnesses, heightened use of invasive vascular procedures and devices, and a growing (albeit still suboptimal) awareness that TE does indeed occur in children. The subsequent portion of this chapter summarizes the clinical spectrum and characterization of pediatric TE, key considerations for diagnostic evaluation and antithrombotic management, and long-term outcomes. Emphasis is also placed on unresolved and emerging clinical and investigative issues in the field.
Characterization of Thromboembolism in Children
TE is anatomically classified by vascular type (i.e., venous versus arterial), distribution (e.g., distal lower extremity versus proximal lower extremity versus central; superficial versus deep vasculature), and organ system affected, if applicable (e.g., renal vein thrombosis, cerebral sinovenous thrombosis, pulmonary embolism [PE]). TE is also distinguished by the additional clinically relevant factors of first episode versus recurrent, symptomatic versus asymptomatic, acute versus chronic (a sometimes-difficult distinction), veno-occlusive versus nonocclusive, and idiopathic versus risk associated. This last category includes both clinical prothrombotic risk factors (e.g., exogenous estrogen administration, indwelling central venous catheter, reduced mobility) and blood-based thrombophilic conditions (e.g., transient or persistent antiphospholipid antibodies, acquired or congenital anticoagulant deficiencies, the factor V Leiden or prothrombin 20210 mutations), as discussed in greater detail later in the section “Etiology.” In light of the frequency of indwelling central venous catheters as a clinical risk factor for TE in children, provoked TE is often also classified as catheter related versus non–catheter related.
Venous Thromboembolism
Epidemiology
Several national or international registries in the past two decades have evaluated the occurrence of venous thromboembolism (VTE) in children. From these data a cumulative incidence of 0.07 per 10,000 (5.3 per 10,000 hospitalizations) was estimated for extremity deep venous thrombosis (DVT) and PE among non-neonatal Canadian children, and an incidence rate of 0.14 per 10,000 children per year was reported for all VTE in the Netherlands. In 2004 evaluation of the National Hospital Discharge Survey and census data for all VTE in the United States disclosed an overall incidence rate of 0.49 per 10,000 per year. More recently, an analysis of the Pediatric Health Information System database revealed a nearly 70% increase in the diagnosis of VTE among hospitalized children, as defined by International Classification of Disease-9 diagnostic codes, over a 7-year period from 2001 to 2007.
Closer examination of epidemiologic data reveals that the age distribution of the incidence rate for VTE in children is bimodal, with peak rates in the neonatal period and adolescence. With regard to the newborn period, the cumulative incidence of venous or arterial TE was reported to be 0.51 per 10,000 births in Germany and 24 per 10,000 admissions to neonatal intensive care units in Southern Ontario. The Dutch registry indicated a VTE-specific incidence rate of 14.5 per 10,000 per year in the neonatal period, approximately 100 times greater than the overall rate in childhood. Among adolescents 15 to 17 years of age, the VTE-specific incidence rate in the United States was determined to be 1.1 per 10,000 per year, a rate nearly threefold that observed overall in childhood.
Although differing selection criteria have contributed to the considerable variability in occurrence estimates in the preceding example, it is clear that the incidence of VTE in children is lower than that among middle-aged and elderly adults. Nevertheless, as discussed later in this chapter, the sequelae of VTE in children, particularly with regard to the post-thrombotic syndrome (PTS), appear to be at least as frequent and severe as those among adults.
Etiology
The pathogenesis of VTE can be readily appreciated by considering Virchow’s triad, consisting of venous stasis, endothelial damage, and the hypercoagulable state. In children, greater than 90% of VTE are risk-associated (compared with approximately 60% in adults), with risk factors often disclosed from more than one component of this triad. Specific examples of VTE risk factors in children are shown in Figure 35-1 . Among the most common of clinical prothrombotic risk factors in childhood is an indwelling central venous catheter. Over 50% of cases of DVT in children and over 80% of cases in newborns occur in association with central venous catheters. The reported cumulative incidence or prevalence of catheter-related thrombosis (CRT) in children receiving home TPN ranges widely, from 1% to 80% ; this broad variation is largely influenced by differing study designs, selection criteria, and diagnostic imaging modalities. The presence of an indwelling central venous catheter, the underlying malignancy or disorder for which bone marrow transplantation was undertaken, and congenital cardiac disease and its corrective surgery were all highly prevalent in the Canadian pediatric thrombosis registry, whereas underlying infectious illness and the presence of an indwelling central venous catheter were identified as pervasive clinical risk factors in a recent cohort study analysis from the United States. It is likely that differences in the composition of referral populations strongly contribute to differences in the composition of VTE etiologies across major pediatric thrombosis centers.
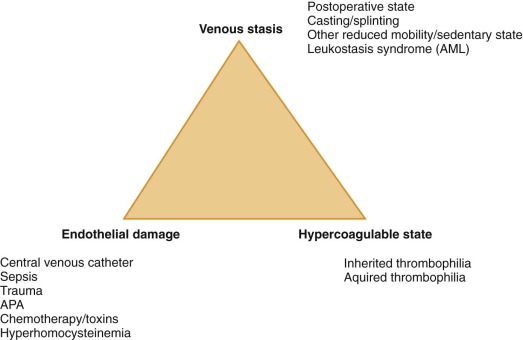
With regard to blood-based risk factors for VTE, thrombophilic conditions (e.g., severe anticoagulant deficiencies , antiphospholipid antibodies [APAs]) in children are frequently acquired, and severe inherited thrombophilias are rare. By contrast, mild congenital thrombophilic traits (e.g., the factor V Leiden and prothrombin 20210 mutations) are common in Caucasian children, as in adults (see Chapter 33 ). Thrombophilia is potentially caused by any alteration in hemostatic balance that increases thrombin production, enhances platelet activation or aggregation, mediates endothelial activation or damage, or inhibits fibrinolysis. Common examples of acquired thrombophilia in children include increased factor VIII activity with significant infection and inflammatory states, anticoagulant deficiencies resulting from consumption in bacterial sepsis and DIC, the production of inhibitory antibodies in acute viral infection, and parainfectious development of APA. To provide an appreciation of the magnitude of VTE risk increase associated with several congenital or genetically influenced thrombophilia traits, population-based VTE risk estimates derived from the adult literature are shown in Table 35-1 . For example, the addition of standard-dose estrogen oral contraceptive pill to underlying heterozygous factor V Leiden substantially increases the risk for VTE from a baseline risk of 15 per 10,000 U.S. females aged 15 to 17 years per year to a risk of more than 500 per 10,000 (or 5%) per year.
| Trait/Condition | VTE Risk Estimate |
|---|---|
| Hyperhomocysteinemia | 2.5× |
| Prothrombin 20210 mutation, heterozygous | 3× |
| OCP (standard dose estrogen) | 4× |
| Factor V Leiden mutation, heterozygous | 2-7× |
| Factor V Leiden mutation, heterozygous, + OCP | 35× |
| Factor V Leiden mutation, homozygous | 80× |
Clinical Presentation
The degree of clinical suspicion for acute VTE in children should be principally influenced by clinical prothrombotic risk factors and family history of early VTE or other vascular disease elicited on thorough interview, known thrombophilia traits and risk factors, and clinical signs and symptoms. The signs and symptoms of VTE depend on anatomic location and organ systems affected and are influenced by characteristics of veno-occlusiveness and chronicity. The classic manifestation of acute extremity DVT is painful unilateral limb swelling. The lack of other physical examination findings (e.g., Homans sign, the presence of a palpable cord in the popliteal fossa in the clinical evaluation for lower extremity DVT) should not reduce the clinical index of suspicion of DVT. In upper extremity DVT with extension into, and occlusion of, the superior vena cava (SVC), signs and symptoms may include swelling of the neck and face, bilateral periorbital edema, and headache. PE is classically manifested by sudden-onset, unexplained shortness of breath with pleuritic chest pain. When PE is proximal or extensive bilaterally in the distal pulmonary arterial tree, hypoxemia is often demonstrated. Associated right-sided heart failure may manifest with hepatomegaly and peripheral edema. Proximal PE and especially saddle embolus can present with cyanosis or sudden collapse. However, in many cases PE may be asymptomatic or produce only subtle symptoms in children, especially when involving limited segmental branches of the pulmonary arteries and not eliciting a significant pleural reaction. In one retrospective series 14% of affected children had no clinical symptoms attributable to PE. Acute cerebral sinovenous thrombosis (CSVT) may present with unusually severe and persistent headache, blurred vision, neurologic signs (e.g., cranial nerve palsy, papilledema), or seizures. The classic findings in renal vein thrombosis (RVT) are hematuria and thrombocytopenia, sometimes associated with uremia (especially when bilateral). Presenting signs include oliguria (especially when bilateral), and in the neonatal period (the time at which RVT is most common during childhood) a flank mass is often palpable on examination. RVT in older children is often associated with nephrotic syndrome (a risk factor for VTE in general owing to the loss of anticoagulant proteins) and hence may present with associated stigmata of peripheral and periorbital edema when diagnosed at presentation of nephrosis. Thrombocytopenia may be a presenting manifestation not only of RVT but also of intracardiac (e.g., right atrial) thrombosis, especially as a CRT associated with sepsis. Isolated intracardiac thrombosis in association with cardiac surgery or central venous catheter placement is most often asymptomatic. Portal vein thrombosis is one of the most common causes of portal hypertension in children, characteristically presenting with splenomegaly, and is associated with findings of hypersplenism (anemia, neutropenia, and thrombocytopenia); gastrointestinal bleeding at presentation typically signals the presence of gastroesophageal varices. Internal jugular vein thrombosis may manifest with neck pain or swelling, and when part of Lemierre syndrome is classically also associated with fever, trismus, pain on lateral neck rotation, and a palpable mass in the lateral triangle of the neck.
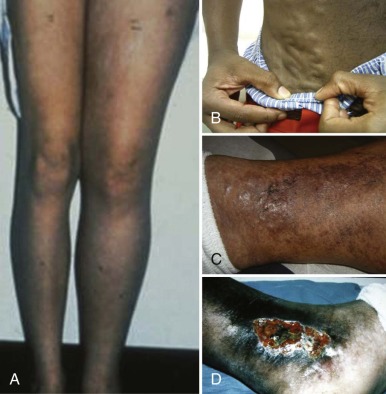
Chronic VTE may be diagnosed incidentally without signs or symptoms (as sometimes occurs for CSVT during unrelated brain imaging), or it may present with signs and symptoms of chronic venous obstruction or PTS, including pain and edema of dependent extremities, dilated superficial collateral veins, venous stasis dermatitis, and frank ulceration of the skin.
Diagnosis
Whereas venography has historically been the gold standard for diagnosis of venous thrombosis, this modality is invasive and has become less prevalent in recent years with the development of effective noninvasive or minimally invasive radiologic imaging technologies. Radiologic imaging is used not only to confirm the clinical diagnosis of VTE but also to define both the extent and occlusiveness of thrombosis. For suspected DVT of the distal or proximal lower extremity, compression ultrasound with Doppler is typically employed for objective confirmation. When the thrombus may affect or extend into deep pelvic or abdominal veins, computed tomography (CT) is often necessary. For circumstances in which radiation exposure is a significant concern or alternative diagnoses (e.g., myositis) are being evaluated, magnetic resonance imaging (MRI) may alternatively be employed. Magnetic resonance venography may be useful in documenting May-Thurner syndrome, an uncommon cause of all lower extremity DVT (but seemingly a common cause of spontaneous or minimally provoked left-sided iliofemoral DVT) in which the iliac vein is compressed by an overriding right common iliac artery. In suspected DVT of the upper extremity, compression ultrasound with Doppler effectively evaluates the limb, but echocardiography or CT is required to disclose involvement of more central vasculature (e.g., right atrial thrombosis). In the case of asymptomatic nonocclusive extremity DVT, venography may be used as an alternative to CT. It is important to recognize that venography (in which radiographic contrast is administered into a vein in the limb) should not be confused with “line-o-grams” (in which radiographic contrast is instilled into a central venous catheter). The latter studies are useful for delineating catheter tip thrombi but cannot reliably detect DVT along the intravascular length of a central venous catheter. Magnetic resonance venography has replaced conventional venography in most centers. However, dynamic conventional venography may be required to evaluate for thoracic outlet syndrome (mechanical compression and entrapment of the subclavian vessels in the region of the thoracic outlet, whether owing to a congenitally narrow subclavian passage between the clavicle, first rib, and a hypertrophied subclavius muscle in athletes [Paget-Schroetter syndrome] or to an accessory rib). To establish a diagnosis of DVT of the jugular venous system, compression ultrasound with Doppler is typically sufficient.
PE in children is commonly diagnosed by spiral CT or, alternatively, by ventilation-perfusion scan, the latter generally being suboptimal in cases wherein other lung pathology exists and at centers wherein availability of, and expertise with, this modality are limited. CSVT is typically diagnosed by standard CT or CT venography, and alternatively by MRI or magnetic resonance venography. The diagnosis of RVT is most often made clinically in neonates and is supported by Doppler ultrasound findings of intrarenal vascular resistive indices; however, in some cases a discrete thrombus may be suggested by Doppler ultrasound, especially when extending into the inferior vena cava. When RVT occurs in older children, Doppler ultrasound or CT is often diagnostic. Similarly, portal vein thrombosis is typically visualized by Doppler ultrasound or CT.
When new-onset venous thrombosis is being evaluated in patients in areas of anatomic abnormality of the venous system (e.g., extensive collateral venous circulation caused by a prior VTE episode, May-Thurner anomaly, atretic inferior vena cava with azygous continuation), more sensitive methods such as CT or magnetic resonance venography are often required to adequately delineate the vascular anatomy as well as the presence, extent, and occlusiveness of thrombosis.
Diagnostic laboratory evaluation for pediatric acute VTE includes a complete blood count, evaluation for thrombophilia (see “ Etiology ”), and beta-human chorionic gonadotropin in postmenarchal women. Additional laboratory studies may be warranted depending on associated medical conditions and VTE involvement of specific organ systems.
Treatment
A summary of conventional antithrombotic agents and corresponding target anticoagulant levels, based on recent pediatric recommendations, is provided in Table 35-2 for both initial (i.e., acute phase) and “extended” (i.e., subacute phase) treatment. Conventional anticoagulants attenuate hypercoagulability, decreasing the risk of thrombus progression and embolism, and rely on intrinsic fibrinolytic mechanisms to break up the thrombus over time. The most commonly employed conventional anticoagulants in children include heparins and warfarin. Heparins, including unfractionated heparin and low-molecular-weight heparin (LMWH), enhance the activity of AT, an intrinsic anticoagulant that serves as a key inhibitor of thrombin. Warfarin acts through antagonism of VK, thereby interfering with γ-carboxylation of the VK-dependent procoagulant factors II, VII, IX, and X.
| Episode | Agents and Target Anticoagulant Activities | Duration of Therapy, by Etiology | |
|---|---|---|---|
| Initial Treatment | Extended Treatment | ||
| First | UFH 0.3-0.7 anti-Xa U/mL | Warfarin INR 2.0-3.0 | Resolved risk factor: 3 months |
| LMWH 0.5-1.0 anti-Xa U/mL | LMWH 0.5-1.0 anti-Xa U/mL | No known clinical risk factor: 6-12 months | |
| Chronic clinical risk factor: 12 months | |||
| Potent congenital thrombophilia: indefinite | |||
| Recurrent | UFH 0.3-0.7 anti-Xa U/mL | Warfarin INR 2.0-3.0 | Resolved risk factor: 6-12 months |
| LMWH 0.5-1.0 anti-Xa U/mL | LMWH 0.5-1.0 anti-Xa U/mL | No known clinical risk factor: 12 months | |
| Chronic clinical risk factor: indefinite | |||
| Potent congenital thrombophilia: indefinite | |||
Initial Anticoagulation.
Initial anticoagulant therapy (i.e., acute phase) for VTE in children generally uses unfractionated or LMWH. LMWH has become increasingly employed as a first-line agent for initial anticoagulant therapy in children, given the relative ease of subcutaneous over intravenous administration, the decreased need for blood monitoring of anticoagulant efficacy, and a decreased risk of the development of heparin-induced thrombocytopenia (HIT). Unfractionated heparin is typically preferred in circumstances of heightened bleeding risk or labile acute clinical status, given the rapid extinction of anticoagulant effect following cessation of the drug. In addition, unfractionated heparin is used for acute VTE therapy in renal insufficiency, given the renal excretion of LMWH. Common initial maintenance dosing for unfractionated heparin in nonneonatal children ranges from 15 to 25 U/kg/h preceded by a loading dose of 50 to 75 U/kg. In full-term neonates a maintenance dose of up to 50 U/kg/h may be required, especially if the clinical condition is complicated by AT consumption. The starting dose for the LMWH enoxaparin in nonneonatal children commonly ranges between 1 and 1.25 mg/kg; no bolus dose is given. In full-term neonates an enoxaparin starting dose of 1.5 mg/kg is typically necessary. For the LMWH dalteparin, initial maintenance dosing of 100 to 150 anti–factor Xa U/kg) appears appropriate based on available pediatric data. Heparin therapy is monitored most accurately by anti–factor Xa activity. For unfractionated heparin the therapeutic range is 0.3 to 0.7 anti–factor Xa activity U/mL, whereas for LMWH the therapeutic range is 0.5 to 1 U/mL. When anti–factor Xa assay is not available, the activated partial thromboplastin time (aPTT) may be used, but it is suboptimal in the pediatric age group, in which antiphospholipid antibodies may interfere with or artifactually prolong the clotting endpoint; indeed, one study of pediatric heparin monitoring demonstrated inaccuracy of aPTT approximately 30% of the time. When dosed by weight in childhood, LMWH does not require frequent monitoring, but anti–factor Xa activity should be evaluated with changes in renal function. In addition, in cases of acute VTE in which acquired AT deficiency is related to consumption in acute infection or inflammation, anti–factor Xa activity may rise as AT levels normalize with resolution of the acute illness; in this circumstance follow-up evaluation of anti–factor Xa activity is warranted in the subacute period. The recommended duration of initial heparin therapy for acute VTE, 5 to 10 days, has been extrapolated from adult data. Unfractionated heparin treatment is rarely maintained beyond the acute period, given the risk of osteoporosis with extended administration and the inconvenience of continuous intravenous administration. Although adult data suggest efficacy of subcutaneous administration of unfractionated heparin for acute VTE, this has been evaluated only for the acute therapy period before extended therapy with warfarin, and the appropriateness of such an approach in children has not been established.
Extended Anticoagulation.
“Extended” anticoagulant therapy (i.e., subacute phase) for VTE in children may employ LMWH or warfarin. Warfarin may be started during the acute phase; however, because severe congenital deficiencies involving the protein C pathway can present as VTE in early childhood and are associated with warfarin skin necrosis, warfarinization should ideally be initiated only after therapeutic anticoagulation is achieved with heparin-based treatment. Warfarin is monitored by INR, derived from the measured PT. The therapeutic INR range for warfarin anticoagulation in VTE is 2 to 3. Historical evidence for maintaining a higher INR (2.5 to 3.5) in the presence of APA is not supported by data from more recent studies ; however, maintaining an INR of 2.5 to 3, when feasible, satisfies both older and more recent literature criteria with regard to appropriate safety and efficacy in this subset of patients.
Duration of Therapy.
Pediatric recommendations for the duration of antithrombotic therapy in acute VTE are largely derived from evidence from adult trials. For a provoked first episode of VTE in children, the recommended duration of anticoagulant therapy is 3 months, as long as the provoking risk factor does not persist beyond 3 months (e.g., postoperative VTE), and—in the authors’ view—absent a potent chronic thrombophilia state (e.g., APA syndrome, homozygous anticoagulant deficiency). The recommended duration is 6 to 12 months when spontaneous and 12 months to lifelong when a chronic risk factor persists (e.g., systemic lupus erythematosus). Recurrent VTE is treated for 6 to 12 months in the presence of an underlying reversible risk factor, 12 months to lifelong when spontaneous, and lifelong when a chronic risk factor persists. Children with SLE and persistence of an LA have a 16- to 25-fold greater risk of TEs than children with SLE and no LA. However, in children with primary (i.e., idiopathic) or secondary (i.e., associated with SLE or other underlying chronic inflammatory condition) VTE, it is possible that the autoimmune disease will become quiescent in later years, such that the benefit of continued VTE prophylaxis may be reevaluated. Some experts have recommended consideration of low-dose anticoagulation as secondary VTE prophylaxis after a conventional 3- to 6-month course of therapeutic anticoagulation for VTE in children with SLE who have APA syndrome. However, further study to optimize the intensity and duration of therapy or secondary prophylaxis for VTE in children with APA syndrome is urgently needed, especially in light of evidence in adult provoked VTE that secondary prophylaxis with low-dose warfarin not only may offer little risk reduction beyond no anticoagulation but also is associated with bleeding complications despite a reduced warfarin dose.
Thrombolytics.
Whereas the aforementioned anticoagulant therapies are conventional in pediatric acute VTE, thrombolytic approaches are gaining increased attention and use during acute VTE therapy in children with hemodynamically significant PE or extensive limb-threatening VTE. Unlike conventional anticoagulants, thrombolytics directly promote fibrinolysis. Tissue-type plasminogen activator is an intrinsic activator of the fibrinolytic system and has been administered exogenously as a systemic bolus or short-duration infusion, a systemic low-dose continuous infusion, or a local catheter-directed infusion with or without interventional mechanical thrombectomy or thrombolysis. A few small cohort study analyses of children with acute proximal limb DVT who had an a priori high risk of poor post-thrombotic syndrome by virtue of completely veno-occlusive thrombus with plasma FVIII activity greater than 150 U/dL or D-dimer concentration greater than 500 ng/mL have revealed that a thrombolytic regimen followed by standard anticoagulation offers potential reduction in the risk of post-thrombotic syndrome when compared (either concomitantly or historically) to standard anticoagulation alone. A National Institutes of Health–sponsored planning grant was begun in 2012 for a future randomized controlled trial on the efficacy and safety of thrombolysis versus standard anticoagulation for acute completely occlusive proximal lower-extremity DVT in children (Goldenberg and Manco-Johnson, personal communication).
Other Anticoagulants.
Other antithrombotic agents include fondaparinux, intravenous and oral direct thrombin inhibitors, and oral direct factor Xa inhibitors. Fondaparinux is a parenteral selective factor Xa inhibitor. It is a synthetic pentasaccharide that rapidly binds to AT in the blood, potentiating the natural inhibitory effect of AT against factor Xa, thus indirectly inhibiting thrombin. Fondaparinux is typically administered once daily as a subcutaneous injection. Dosing in children has been informed by a pediatric pharmokinetic and safety study. Direct thrombin inhibitors, by contrast, inhibit thrombin directly by binding to exosite I or the active site of thrombin (or both). These drugs include hirudin and recombinant hirudins such as lepirudin. Argatroban is a synthetic small molecule derived from L-arginine and functions as a reversible direct thrombin inhibitor. Intravenous direct thrombin inhibitors are routinely monitored by aPTT, with the therapeutic goal ranging from a 1.5- to 3.0-fold aPTT prolongation. At present, intravenous direct thrombin inhibitors are indicated for the treatment of HIT, particularly when associated with acute thrombosis (HITT). Indeed, this is the setting in which pharmacokinetic studies of argatroban have been conducted. However, pediatric use of intravenous direct thrombin inhibitors has also been reported in settings of catastrophic APA syndrome and thrombotic storm.
Oral direct thrombin inhibitors and oral direct factor Xa inhibitors have been developed as a possible alternative to warfarin. They have the advantage of oral bioavailability, low intersubject variation, no metabolism by the hepatic cytochrome P450 system, a low incidence of drug-drug interactions, and no dietary influence. Early-phase pediatric clinical trials in pediatric VTE prevention or treatment with the oral direct thrombin inhibitor dabigatran and the oral direct factor Xa inhibitors apixaban and rivaroxaban were all initiated between 2011 and 2012, after regulatory agency approvals for the corresponding adult indications.
Other Therapeutic Agents.
Other products may have antithrombotic roles in selected circumstances but lack definitive demonstration of efficacy and safety in clinical trials. For example, plasma replacement with protein C concentrate is a useful adjunctive therapy for VTE or purpura fulminans owing to microvascular thrombosis in severe congenital protein C deficiency and may play a beneficial role in the treatment of purpura fulminans owing to microvascular thrombosis in children with sepsis, particularly in meningococcemia. In addition, case series have suggested a role for AT replacement in prevention of VTE in children and young adults with congenital severe AT deficiency, for the prevention of L-asparaginase–associated VTE in pediatric acute lymphoblastic leukemia (ALL), and as combination therapy with defibrotide in the prevention and treatment of hepatic sinusoidal obstruction syndrome (formerly termed veno-occlusive disease ) in children undergoing hematopoietic stem cell transplanation. The potential benefit toward VTE risk reduction of a regimen of AT replacement combined with daily prophylactic LMWH during induction and consolidation phases of therapy in ALL has also been suggested by a historically controlled cohort study of the BFM 2000 protocol experience in Europe. AT replacement may also be worthy of consideration in patients with acute VTE undergoing heparinization in whom significant AT deficiency prevents the achievement of therapeutic anti–factor Xa levels (i.e., heparin “resistance”). This may be seen in neonates with clinical conditions complicated by AT consumption superimposed on the physiologic relative deficiency of this key intrinsic thrombin inhibitor in the newborn period. However, AT replacement in preterm neonates has been investigated to improve the outcomes of respiratory distress syndrome, intracranial hemorrhage, and sepsis but has failed to demonstrate benefit and may contribute to adverse events.
Vena Cava Filters.
In certain circumstances vena cava filters may serve an adjunctive role in antithrombotic management in children. The use of vena cava filters should be considered in children of appropriate size in whom contraindications to anticoagulation exist or in whom recurrent VTE (especially PE) has occurred despite therapeutic anticoagulation and in the absence of a reversible risk factor. In addition, “temporary” or retrievable vena cava filters may be considered during times of especially heightened risk for PE or at times when therapeutic anticoagulation is transiently considered to engender a heightened risk of hemorrhagic complications (e.g., recent neurosurgery). The presence of the filter alone is not an indication for long-term anticoagulation because the risk for vena caval thrombosis related to the device is low. Long-term outcomes in adult series have shown a failure rate (clinically relevant, symptomatic PE) of 3.3% to 5.6%. Major complications were infrequent (<1%) and include rare cases of caudal migration, filter fractures, or caval wall perforation. A retrospective review of 15 children 18 years of age or younger suggested efficacy of long-standing vena cava filters with regard to primary and secondary prevention of PE. Follow-up was a mean of 9.2 years (range 19 months to 16 years), and there were no significant complications, complementing similar findings in other small case series. However, until the impact of such nonretrievable devices on the vena cava of the developing child has been better studied, their use in preadolescents should be undertaken with caution. Experience with surgical removal of retrievable vena cava filters is quite limited in children with unknown risks and costs of extraction. Surgical removal of permanent vena cava filters should be strongly discouraged because this often requires vena caval ligation.
Outcomes
Complications of VTEs can occur both acutely and over the long term and are germane to both CRT and non-CRT. Short-term adverse outcomes include major hemorrhagic complications of antithrombotic interventions, as well as of the thrombotic event itself (e.g., post-thrombotic hemorrhage in the brain or adrenal gland); thrombus progression (involvement of previously unaffected contiguous venous segments, or change from nonocclusive to occlusive); early recurrent VTE (including DVT and PE ); SVC syndrome in DVT of the upper venous system ; acute renal insufficiency in RVT ; catheter-related sepsis, PE, and catheter malfunction (sometimes necessitating surgical replacement) in CRT; severe acute venous insufficiency leading to venous infarction with limb gangrene in rare cases of occlusive DVT involving the extremities; and death from hemodynamic instability in extensive intracardiac thrombosis or proximal PE. Long-term adverse outcomes in pediatric VTE have been previously reviewed and include recurrent VTE; chronic hypertension and renal insufficiency in RVT; variceal hemorrhage in portal vein thrombosis ; chronic SVC syndrome in CRT involving SVC occlusion; loss of availability for venous access in recurrent or extensive CRT of the upper venous system; and development of PTS, a condition of chronic venous insufficiency following DVT. The manifestations of PTS include (in order of increasing severity) edema, visibly dilated superficial collateral veins, venous stasis dermatitis, and venous stasis ulcers ( Fig. 35-2 ). Given the long-term and sometimes lifelong risks of disease sequelae as in RVT and functional impairment as in PTS, venous thrombosis is best considered a chronic disorder in children.