2 Department of Neurology, Thomas Jefferson University, and Jefferson Medical College, Philadelphia, PA, USA
3 Department of Pediatric Neurology and Developmental Medicine, University Children’s Hospital, Tübingen, Germany
MLD and GLD
Metachromatic leukodystrophy (MLD) and globoid cell leukodystrophy (Krabbe disease, GLD) are autosomal recessively inherited disorders caused by the deficiency of arylsulfatase A (ASA) and galactosylceramidase (GALC), respectively. Since both enzymes depend on the assistance of activator proteins saposin B (ASA) and A (GALC), respectively, clinically indistinguishable diseases can also be caused by the deficiencies of these activator proteins (Figure 9.1). The pathological hallmark of both diseases is a loss of oligodendrocytes and thus myelin leading to multiple progressive and finally lethal neurologic symptoms [1, 2].
Case studies
Figure 9.1 Enzymes and activator proteins involved in degradation of sulfatide and galactosylceramide. Sulfatide is hydrolyzed by arylsulfatase A (ASA) to yield galactosylceramide and sulfate. The activator protein saposin B solubilizes sulfatide and presents it to the enzyme. Sulfatide accumulates in the absence of either protein. This step is defective in metachromatic leukodystrophy (MLD). Galactosylceramide is cleaved into galactose and ceramide by galactosylceramidase (GALC). This enzyme depends on the assistance of saposin A. A deficiency of either of these proteins causes globoid cell leukodystrophy (GLD) (Krabbe disease).
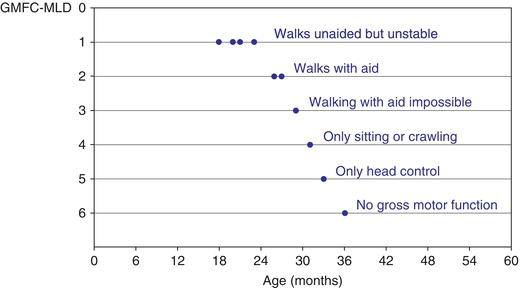
Figure 9.2 Natural course of MLD. Typical course of gross motor function in a child with late-infantile MLD (see case history oflate-infantile MLD) described by a gross motor function classification, the GMFC-MLD [9] : first abnormalities at 18 months (walksunstable); once level 2 (walking only with aids) is attained, gross motor function rapidly deteriorates and at the age of 3 years, there isno gross motor function anymore.
Epidemiology
In the European population the incidence of MLD and GLD is estimated to be about 0.6 in 100,000 live newborns for each disorder [3]. Activator protein deficiencies are much rarer. Their incidence has not been exactly determined. In general, the diseases occur in all ethnicities. Higher incidences of MLD are found in Habbanite Jews (1:75), Christian Arabs (1:8,000), Eskimos and Navajo Indians. A higher frequency of GLD has been described in certain Druze and Arab villages in Israel (1:170) [4]. To initiate therapy before symptoms are obvious, New York State instituted a newborn screening test for Krabbe disease using dried blood spots in August 2006. Since that time over 1.2 million newborns have been tested, and 4 were found to have infantile Krabbe disease based on a combination of mutation analysis and follow-up enzyme testing.
Genetics
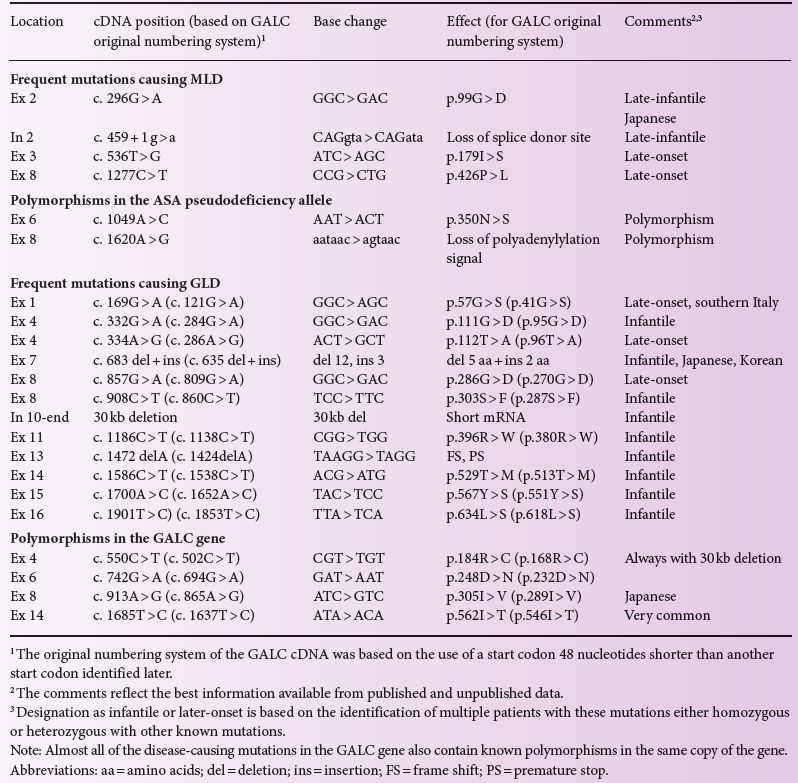
The genes for ASA and GALC are located on 22q13.31-qter (ASA) and 14q31 (GALC), respectively. Whereas the ASA is a small gene in which the 8 exons encompass only about 3 kb of genomic sequence, the 17 exons of the GALC gene span 56 kb.
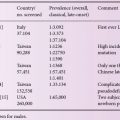
More than 150 mutations are known in the ASA gene. Among Caucasians, only a few of them are frequent (c.459 + 1 g > a, p.426P > L, p.179I > S) accounting for ~ 60% of all alleles, whereas many of the other mutations were only found in a few, mostly single, families (Table 9.1). More than 110 mutations in the GALC gene are known. One mutation (p.57 G > S) in the GALC gene is common in southern Italy. Japanese patients display a different spectrum of genetic defects in both disorders. In the ASA gene this is a p.99 G > R substitution and for the GALC gene it is a deletion/insertion mutation c.683_694del12insCTC. Table 9.1 summarizes the most common disease-causing mutations found in patients with MLD and GLD.
In both diseases there is a limited genotype/phenotype correlation. In MLD, homozygosity for mutations which do not allow the synthesis of any functional enzyme, always cause the severe late infantile form of disease. The presence of one allele associated with residual enzyme activity mitigates the disease to the juvenile form, whereas homozygosity for two alleles with residual ASA activity in most cases will result in a late juvenile or even adult form of MLD (Figure 9.3). Thus, residual enzyme activity which, in late-onset patients, is in the range of 2–4% of normal, is one determinant of clinical severity. However, clinical variability in late-onset patients – even in siblings with identical ASA genotype – is considerable, showing that other genetic or epigenetic factors influence the disease course substantially. Therefore, a precise prediction of the individual course of disease based on genotype analysis is impossible.
Data on genotype/phenotype correlations in GLD are more limited than in MLD, but the data available suggests that correlations in GLD and MLD follow similar rules. The finding of certain mutations in the GALC gene, either homozygous or heterozygous with known mutations, can predict a severe phenotype in a newborn individual. The presence of other known mutations in the GALC gene will predict a later-onset form of GLD. Late-onset MLD patients frequently present initially with psychiatric symptoms rather than motor problems. This phenotype correlates with a certain genotype. Patients presenting psychiatrically are frequently heterozygous for a null allele and the p.179I > S amino acid substitution [5]. The molecular basis for this peculiar genotype/phenotype correlation remains unclear.
Table 9.1 Most common mutations and polymorphisms found in patients with MLD and GLD.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


