2 Department of Neurology, Pediatrics and Medical Genetics, Mayo Clinic, Rochester, MN, USA
Niemann–Pick disease type C [NPC] (Online Mendelian Inheritance in Man (OMIM)# 257220 – NPC1, OMIM# 607625 – NPC2) is a rare autosomal recessive metabolic disorder caused by the deficient function of either of two proteins, named NPC1 and NPC2, which act cooperatively to facilitate the egress of free cholesterol and other compounds from the endolysosomal system. The impaired activity of these proteins is associated with the accumulation of free cholesterol and a variety of sphingolipids in the lysosomes but their precise function(s) have not yet been fully elucidated [1–3].
Case histories
Epidemiology
Niemann–Pick disease type C has a calculated incidence of 1:100,000–120,000 live births [3, 4]. It is likely more common than this number suggests, as adult onset cases are being recognized more often, and as awareness of the wide range of presentations of this disease grows among physicians. All ethnic groups are affected; genetic isolates with an increased carrier frequency and incidence have been described in the Acadian descendants of Jean Amirault in Nova Scotia, in a Hispanic group in New Mexico and Colorado and in a Bedouin group [1].
Genetic basis
Mutations in the NPC1 gene, which is located at 18q11-q12, are present in about 95% of patients with NPC. More than 300 disease-causing mutations (and numerous polymorphisms) have now been identified (about one-third of which are missense mutations; the remainder comprise frameshift and nonsense mutations, single amino acid deletions and insertions, splice mutations and large deletions) [5]. Only the p.I1061T and p.P1007A mutations are globally frequent and account together for close to 20% of mutated alleles in several countries. An increasing number of recurrent mutant alleles are being described, but a majority of mutations are “private”. Twenty different mutations in the NPC2 gene (located at 14q24.3) have been described in about 40 families. NPC1 and NPC2 mutations correlate with the neurological form of the disease, not with the systemic manifestations.
Pathophysiology
The pathophysiology of NPC is incompletely understood. The microscopic and macroscopic consequences of NPC1 and NPC2 mutations are well documented: variable enlargement of the liver and spleen, with foam cell infiltrates and sea blue histiocytes there and in the bone marrow. In the brain, neurons are ballooned with stored lipids, and ectopic dendritogenesis, meganeurite formation and neuroaxonal dystrophy are characteristic. A major feature is progressive neuronal death, particularly of Purkinje cells. Neurofibrillary tangles are seen in the cortex [1]. Biochemical analysis shows marked storage of unesterified cholesterol, sphingomyelin, bis(monoacylglycero)phosphate, free sphingoid bases (sphingosine and sphinganine) and several glycosphingolipids in peripheral tissues (especially spleen), with mostly increased glycosphingolipids in the brain [1, 3].
The sequence of events that leads to these tissue changes is not known, but current theories centre around cholesterol, free sphingoid bases and glycosphingolipid accumulation as the initiating events. The “classical” model stresses the central role of NPC1 and NPC2 proteins in the trafficking of cholesterol in the late endosomal/ lysosomal system [6], and infers that cholesterol accumulation further impairs trafficking and leads to the accumulation of other lipid species as a secondary event. Based on in vitro studies indicating that free sphingosine is the first species to accumulate in NPC1 fibroblasts, a challenging model has been proposed, where sphingosine accumulation leads to abnormal intracellular distribution of calcium that in turn triggers accumulation of cholesterol and sphingolipids [7]. That NPC2 and NPC1 are able to bind and transfer free cholesterol in a sequential fashion seems fairly well established [6]; secondary effects of the lysosomal storage of cholesterol on trafficking of sphingolipids have also been described. Similarly, the potential deleterious effects of abnormally high levels of free sphingoid bases or lysosphingolipids have been documented. Considering the severity of NPC in the nervous system, an apparently intriguing point is the limited accumulation of cholesterol seen in neurons, although this can probably be explained by the specifics of cholesterol metabolism in the brain [8]. Of note is the fact that the increase in free sphingosine is also small in the brain, where the most conspicuous accumulation is that of ganglioside GM2. Interestingly, early and repeated intrathecal administration of cyclodextrin to the cat model normalized the levels of all three lipids. Thus, whatever the mechanism, increases of cholesterol, sphingosine and ganglioside appear to be closely linked. More work is needed to better understand the brain dysfunction in NPC, particularly to identify the trigger (or triggers) for patterned neuronal loss.
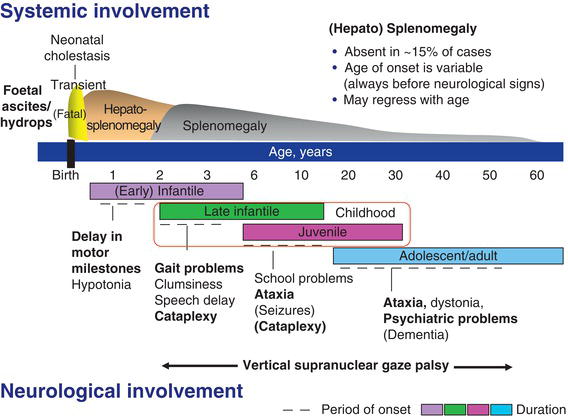
Figure 11.1 Schematic representation of the clinical aspects of Niemann–Pick C disease. (Modified from Vanier [3].)
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


