Fig. 3.1
Cancer vaccination and induction of T cell activation
Vaccine Platforms and Vectors
Common Mechanisms of Action
This section will discuss various therapeutic cancer vaccine constructs, including peptide, DNA, whole tumour cell, vector-based, and ex vivo processed dendritic cell (DC) vaccines. All vaccines require dendritic cell activation with presentation of the target antigen via major histocompatibility (MHC) complexes (signal 1) and a costimulatory signal (signal 2) (Fig. 3.2) [1]. While this final step of activated DCs inducing T cell activation is common, each vaccine platform has a unique method of DC stimulation. For instance, a peptide vaccine is composed of a target peptide, which represents the target antigen overexpressed on the tumour cell via MHC class 1 and another chemical, called an adjuvant, which has the role of stimulating DC maturation toward an activated state. On the other hand, vector based platforms may trigger DC activation via the presence of the foreign vector, as in the case of a yeast-based vaccine platform. In each case, the vaccine design must include a method of delivering the target antigen for DC uptake and presentation while also containing a component of the innate immune response that will induce a “danger signal” [2] and drive the DC to activate the adaptive immune response [3, 4]. Selection of the target antigen, the concentration and dwell time of the vaccine, along with the concentration of administered adjuvant are all critical to driving an effector T cell phenotype necessary for anti-tumour activity [5].
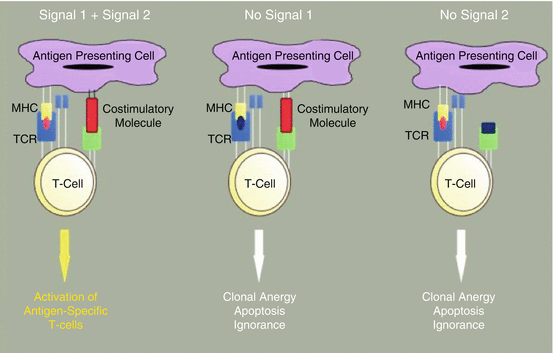
Fig. 3.2
T cell dependence on co-stimulation
Peptide Vaccines (Table 3.1)
Table 3.1
Peptide vaccine clinical trials
Vaccine | Cancer type | Trial design | n | Treatment arms | mOS |
|---|---|---|---|---|---|
Mutant RAS | |||||
GI-4000 [13] | Pancreatic | Adjuvant, phase II | 176 | 3 weekly doses of GI-4000 vs. placebo, followed by 6 cycles adjuvant gemcitabine | 17.5 vs. 14.8 months |
CTN-95002 CTN-98010 [171] | Pancreatic | Adjuvant, phase I/II | 23 | 6 administrations on weeks 1, 2, 3, 4, 6, and 10 | 27.5 months |
CEA | |||||
CEAVAC (3H1) + TriAb (CALGB89903) [24] | Colorectal | Resected metastatic, adjuvant, phase II | 56 | 4 administrations every 2 weeks for 2 months, then monthly for 2 years, and then every 2 months for 3 years | Not reached (2 year OS: 94 %) |
CEAVAC + TriAb [23] | Colorectal | Metastatic, phase III | 630 | 5-FU/leucovorin plus CEAVac vs. placebo | 21.3 vs. 18.5 months (in patients receiving ≥8 cycles) |
β-hCG | |||||
CTP37-DT [43] | Colorectal | Advanced, phase II | 77 | 3 administrations on days 0, 28, and 70 | 10.5 vs. 5.5 months (high- vs. low-responders) |
Avicine (conjugated to DT) [44] | Pancreatic | Advanced, phase II | 55 | Gemcitabine plus Avicine vs. gemcitabine alone | 6.6 vs. 4.7 months |
Gastrin | |||||
G17DT [58] | Pancreatic | Advanced, phase II | 30 | 3 administrations on weeks 0, 2, and 6 | 6.2 months (all), 7.2 vs. 4.0 months (high- vs. low-responders) |
G17DT [60] | Gastric | Untreated unresected or metastatic, phase II | 103 | Cisplatin, 5-FU, and G17DT | 9.0 months |
G17DT [61] | Pancreatic | Advanced, phase II | 154 | G17DT vs. placebo | 5.0 vs. 2.7 months |
G17DT [62] | Colorectal | Metastatic, progressed on irinotecan, phase II | 161 | G17DT plus irinotecan | 9.0 vs. 5.6 months (immune responders vs. non-responders) |
CD55 | |||||
105 AD7 [67] | Colorectal | Advanced, phase II | 162 | 105 AD7 vs. placebo | 4.1 vs. 6.1 months |
CD17-1A | |||||
CD17-1A [78] | Colorectal | Metastatic, phase IB/II | 20 | CD17-1A plus GM-CSF plus IL-2 | 8.0 months |
Edrecolomab [80] | Colorectal | Adjuvant (stage III), phase III | 2761 | 5-FU/leucovorin plus edrecolomab vs. 5-FU/leucovorin alone vs. edrecolomab alone | Not reached (3 year OS: 74.7 % vs. 76.1 % vs. 70.1 %) |
Telomerase | |||||
GV1001 [82] | Pancreatic | Unresectable, Phase I/II | 48 | Low-, intermediate-, and high-dose groups | 4.0 vs. 8.7 vs. 5.1 months |
GV1001 (TeloVac) [84] | Pancreatic | Advanced, Phase III | 1062 | Gemcitabine and capecitabine, gemcitabine and capecitabine followed by GV1001, or concurrent | 7.9 vs. 6.9 vs 8.4 months |
WT1 | |||||
WT1 [85] | Pancreatic | Advanced, phase I | 32 | Gemcitabine plus WT1 vaccine | 10.9 vs. 3.9 months (immune responders vs. non-responders) |
WT1 [86] | Pancreatic or biliary | Advanced, phase I | 25 | Gemcitabine plus WT1 vaccine | 9.6 months (biliary) and 8.6 months (pancreatic) |
Mutant RAS
RAS oncogenes are associated with a variety of malignancies and are mutated in many GI cancers. RAS proteins function as GTPases that playing critical roles in signal transduction pathways, serving as an on/off switches and promoting cell proliferation when switched on [6]. Mutated RAS genes—KRAS, HRAS, and NRAS—code for altered RAS proteins, which lead to constitutive activation of the downstream signalling pathways [7]. RAS mutation status conveys vital clinical information, guiding therapeutic selection in metastatic colorectal cancer (CRC); In particular, cetuximab and panitumumab have decreased efficacy in tumours with RAS mutations due to circumvention of EGFR inhibition [8, 9]. These mutations are extremely common in pancreatic ductal adenocarcinoma (approximately 90 % of tumours harbour a KRAS mutation, the majority of which are in codon 12, with others in codons 13 and 61) [10, 11]. Given that RAS mutations carry a pivotal role in oncogenesis, RAS vaccines have been developed to selectively target GI cancers.
One such RAS vaccine is GI-4000, which has been studied in colorectal, pancreatic, and non-small cell lung cancers (NSCLC) [12]. GI-4000 is comprised of four different strains of heat-inactivated Saccharomyces cerevisiae yeast expressing the 7 most common RAS mutations; each strain has a fusion protein of three different RAS mutations (GI-4014: Q61L, G12V, and Q61R; GI-4015: Q61L, G12C, and Q61R; GI-4016: Q61L, G12D, and Q61R; and GI-4020: Q61L, G12R, and Q61H) [13]. In an adjuvant phase II trial, 176 patients with RAS mutant pancreatic cancer status-post resection were randomised 1:1 to receive 3 weekly doses of GI-4000 or placebo followed by six cycles of gemcitabine (1000 mg/m2 IV on days 1, 8, and 15 every 21 days) with monthly GI-4000 or placebo administered during off-weeks. Patients were stratified by resection status (R0 or R1).
Among 39 patients who had R1 resections, those who received GI-4000 had a median overall survival (mOS) of 524 days versus 444 days in patients who received placebo [13]. In addition, patients treated with GI-4000 had a higher mutation-specific T cell response (determined by enzyme-linked immunosorbent spot (ELISPOT) assay [serum interferon γ analysis, 46.7 % vs. 8.3 %, P = 0.032]). Accordingly, GI-4000 treated immune responders had the greatest survival advantage (mOS = 596 days vs. 444 days with placebo).
Among patients who had R0 resections, those with G12R mutations had better mOS irrespective of receiving GI-4000 or placebo (335 days longer compared with other mutations), and these patients also benefited from GI-4000 over placebo (mOS was 568 days longer for patients treated with GI-4000) [14]. This finding may be attributed to a significant decrease in inhibitor T regulatory cells (Tregs; 26.2 % of patients treated with GI-4000 vs. 8.8 % treated with placebo had at least a 2-fold decrease in Tregs, P = 0.048). Given these mixed results, phase III studies of GI-4000 have not been performed.
A retrospective analysis looked at 20 evaluable patients with resected pancreatic adenocarcinoma treated with 2 Norwegian KRAS peptide vaccines on 2 prior adjuvant phase I/II trials (CTN-95002, 4 different peptides of KRAS 5-21 corresponding to the patient’s KRAS mutation, and CTN-98010, a mixture of 7 peptides of KRAS 5-21). Seventeen of the 20 patients had an immunologic response to the vaccine, based on positive delayed-type hypersensitivity (DTH) test or presence of KRAS-specific T cells in the peripheral blood after vaccination. The mOS for all patients was 27.5 months, the 5-year survival rate was 22 % (29 % in immune responders), and 20 % of patients were alive at 10 years. Although this was a retrospective analysis involving only a small number of patients, the study still manages to demonstrate the potential for a durable immune responses to RAS peptide vaccines.
CEA (See Also Section “Vector-Based Vaccines”)
Carcinoembryonic antigen (CEA) is a glycoprotein involved in cell adhesion, which is normally produced during fetal development. It is also a tumour-associated antigen that is expressed on over 90 % of colorectal and pancreatic adenocarcinomas, 70 % of NSCLC, and 50 % of breast cancers [15–17]. Given its frequent presence on the surface of tumour cells, there have been many attempts to incorporate CEA into vaccines. CEA by itself is poorly immunogenic due to immune tolerance, although cytotoxic T-lymphocytes (CTLs) recognise CEA epitopes that bind to major histocompatibility complexes (MHC) A2, A3, and A24 [18]. Further modification of the HLA-A2 CEA CAP-1 epitope to CAP1-6D has been shown to increase its immunogenicity, enhancing sensitisation of CTLs by 100–1000 fold [19, 20].
Foon et al. [21] first attempted to use an anti-idiotype antibody that is the internal image of CEA (3H1, the precursor to CEAVac, given subcutaneously at a dose of 1, 2, or 4 mg every 2 weeks for 4 injections, and then monthly thereafter) in a phase Ib trial of 23 patients with metastatic CEA-positive CRC. Seventeen patients developed antibodies against the vaccine, indicating an immune response, and the presence of this response correlated with progression free survival (PFS) and overall survival (OS), although disappointingly, there were no objective clinical responses. These findings led to a single-arm phase II trial of CEAVac at the 2 mg dose in 32 patients with resected and incompletely resected CRC (14 patients were also treated concurrently with 5-fluorouracil (5-FU) chemotherapy regimens) [22]. All 32 patients generated high-titre antibody responses (based on increased IgG to CEA), and these were not affected by co-administration with 5-FU. A phase III trial of 630 patients with stage IV CRC randomised patients receiving 5-FU/leucovorin to CEAVac versus. placebo. Whereas over 75 % of patients in the CEAVac arm developed anti-CEA antibodies, there was no difference in mOS between the treatment arms, although there was a significant difference in patients who received at least 8 cycles of treatment (21.3 vs. 18.5 months with placebo, P = 0.04) [23].
In the phase II Cancer and Leukaemia Group B (CALBG) 89903 study, 52 patients with resected metastatic colorectal cancer (mCRC) to the liver received 4 biweekly treatments of anti-idiotype monoclonal antibodies against CEA (CEAVac) combined with human milk fat globule (TriAb), followed by monthly treatments for 2 years and then every other month for 3 years. Although the treatment was well tolerated with only 10 % of patients experiencing a grade 3 adverse event (AE), the recurrence-free survival rate at 2 years was 39 %, which was not an improvement on the expected rate of 40 % following hepatic resection alone [24].
Following suggestions that tumour antigen peptide emulsification in Montanide adjuvant combined with granulocyte-macrophage colony-stimulating factor (GM-CSF) could stimulate DC differentiation and improve DC recruitment, a phase I study of a modified CEA peptide (CAP1-6D)/montanide/GM-CSF-vaccine was conducted. All patients included in the study had pancreatic adenocarcinoma expressing CEA and HLA-A2, which had been previously treated. Nineteen patients were randomised to receive 3 different doses of the vaccine every 2 weeks at doses of 10 μg, 100 μg, or 1000 μg. The CTL response (measured by ELISPOT testing) was dose-dependent, with 248 spots per 10 [4] CD8 cells following a 1000 μg dose, compared with 37 spots following a 10 μg dose (P = 0.037) [25].
Although this study demonstrated a strong immune response to the CAP1-6D/montanide/GM-CSF-vaccine, along with limited toxicity, it has not been further studied.
MUC-1
Mucin 1 (MUC-1) is a transmembrane protein with anti-adhesion properties that may promote metastasis and is expressed on many adenocarcinomas [26]. Thus, MUC-1 is of interest as a tumour-associated antigen for use in therapeutic vaccines. In an early phase I trial, 63 patients (including 24 with pancreatic cancer and 30 with CRC) were treated with a synthetic mucin peptide mixed with the bacillus calmette-guérin (BCG) vaccine [27]. Seven of 22 patients tested had a two to four-fold increase in mucin-specific CTL, but there were no objective tumour responses and only three patients had stable disease. Other early studies used a fusion protein with MUC-1 conjugated to mannan. Karanikas et al. [28], gave four to eight subcutaneous injections of MUC-1/mannan fusion protein to 25 patients with advanced cancers—16 with colorectal, one with gastric, and 8 with breast cancers—on weeks 1, 2, 3, 4, 7, 9, 11, and 13 at escalating doses (10–500 μg per injection). Thirteen of the 25 patients had a detectable antibody response by ELISA, and CTL responses were seen in 2 out of 10 evaluable patients. In subsequent phase I trials, an intraperitoneal route of administration tended to amplify immune response, but the use of cyclophosphamide to increase cellular immunity was found to be of no benefit [29].
Kimura et al. [30] published the results of a phase I/II immunoprevention study, in which 39 patients with premalignant colonic adenomas were given a MUC-1 peptide vaccine with a toll-like receptor (TLR)3 agonist (polyinosinicpolycytidylic acid stabilised with poly-L-lysine and carboxymethylcellulose, poly-ICLC) on weeks 0, 2, and 10. Seventeen of the 39 vaccinated patients achieved an immune response, manifested by high anti-MUC-1 immunoglobulin G (IgG) levels with lasting immune memory—12 of 16 patients who responded at week 12 also responded to a booster injection at week 52. Those patients who did have an immune response tended to have higher levels of myeloid-derived suppressor cells (MDSCs) prior to vaccination. This prevention strategy is being further evaluated in an ongoing randomised, double-blind, placebo-controlled phase II trial (NCT02134925).
SART3
Squamous cell carcinoma antigen recognised by T cells 3 (SART3) is a tumour-rejection antigen that is expressed in over 70 % of CRCs. SART3 possesses two antigenic epitopes (SART3109-118 and SART3315-323), and can induce CTLs in HLA-A2 and HLA-A24 positive patients [31–34]. In a phase I study, Miyagi et al. [35], administered a vaccine containing the two SART3 antigenic peptides mixed with incomplete Freund’s adjuvant to 12 patients with advanced CRC (all were refractory to 5-FU-based chemotherapy except for 1 patient who was treatment naïve; all patients were confirmed to be HLA-A24 positive). At 5 weeks, 9 patients had stable disease and 2 had progressive disease (one was not evaluable); a CTL response was seen in 7 out of 11 evaluable patients. Tit was found that the CTL response did not correlate with improved outcomes, and no specific IgG or immunoglobulin E (IgE) responses to the peptides were detected. No further clinical trials using SART3 peptide vaccines have been performed.
β-hCG
Human chorionic gonadotropin (hCG) is a glycoprotein hormone composed of α and β subunits. β-hCG is used as a marker for pregnancy, although it is also elevated in trophoblastic and non-trophoblastic malignancies [36, 37]. In patients with CRC, wide ranges of serum hCG levels (2–41 %) and tumour hCG expression (17–52 %) have been reported, and both have been shown to have a negative prognostic significance [38, 39]. Importantly, β-hCG is not expressed on normal colonic mucosa or benign colonic lesions [40–42]. In a randomised phase II trial, Moulton et al. [43], studied CTP37-DT, a synthetic β-hCG peptide vaccine conjugated to diphtheria toxoid (DT), in 77 patients with advanced CRC, randomised into low-dose and high-dose groups. The vaccine was administered on days 0, 28, and 70. It was found that 73 % of patients produced anti-hCG antibodies, and those with an antibody titre above the median value had improved survival compared with those with a titre below the median (44.9 vs. 23.6 weeks, P = 0.0002). There were no statistically significant differences between the low- and high-dose groups in terms of antibody response, survival, and toxicity.
Another phase II study randomised 55 patients with treatment-naïve advanced pancreatic cancer to receive gemcitabine or gemcitabine plus Avicine, a vaccine composed of two synthetic β-hCG peptides conjugated to DT. All immunised patients produced anti-β-hCG antibodies and mOS was improved in this treatment cohort, compared with those receiving gemcitabine alone (6.6 vs. 4.7 months; 1-year survival 30 % vs. 14 %) [44]. Despite this result, no further investigations of peptide-based β-hCG vaccines have been published.
Survivin
Survivin is a member of the inhibitor of apoptosis family of proteins, and functions by inhibiting caspase activation and preventing programmed cell death [45, 46]. Survivin is expressed in fetal tissue and many tumours, but is absent in normal, fully differentiated cells, making it a potential target for cancer vaccines [47, 48].
Hirohashi et al. [49], identified survivin-2B80-88, an HLA-A24-restricted peptide recognised by CD8+ CTLs. Survivin-2B80-88 was subsequently studied in combination with incomplete Freund’s adjuvant (IFA) and type-1 interferon α (IFN) for treatment of advanced CRC [50, 51] and pancreatic cancer [52]. In the CRC study, two groups of patients were treated. The first group of five patients were treated with peptide and IFA only. The second group of eight patients were treated with peptide, IFA, and IFN. One patient in the first group had stable disease, while the other four in this group had progressive disease. Out of the eight patients in the second group, four had stable disease, while the other four had progressive disease [51]. Patients in the IFN group also had an increase in peptide-specific CTL clones by tetramer staining, indicating an enhancement in immunogenicity. The pancreatic cancer study similarly treated patients with peptide, IFA, and IFN, and four out of six patients treated with this combination displayed stable disease. The same four patients also had evidence of an immunologic response by tetramer staining and ELISPOT assay [52].
Finally, a phase I study by Lennerz and colleagues [53] used EMD640744, a combination of 5 HLA class I-binding survivin peptides, and administered this with the adjuvant MontanideTM ISA 51 VG to 49 cancer patients, including 10 patients with CRC. Considering all 49 patients, the best overall response was stable disease (seen in 28 % of patients), and 63 % of patients had peptide-specific CTL immunologic responses by multimer staining and ELISPOT testing [53].
While survivin-based peptide vaccines have clearly demonstrated an ability to elicit peptide-specific immune responses (especially with the addition of IFN), their use has not progressed beyond early phase clinical trials given their modest clinical responses.
G17DT
G17DT is an immunoconjugate of gastrin-17 linked to DT. It is administered intramuscularly, inducing antibody formation against gastrin-17 and cell-associated precursor gastrin molecules [54]. Gastrin functions as a growth peptide in both colorectal [54, 55] and pancreatic cancers [56, 57]. Brett et al. [58], studied the vaccine in 30 patients with advanced pancreatic cancer as part of a phase II study. Tweinty vaccine-treated patients (approximately 67 %) achieved an antibody response, and those exposed to higher dose level had a higher response rate (82.4 % in the group receiving 250 μg G17DT vs. 46.2 % in the 100 μg group, P = 0.018). mOS was significantly longer in antibody responders compared with non-responders (217 days vs. 121 days, P = 0.0023).
Another phase II study of G17DT in 52 patients with stage I-IV gastric adenocarcinoma also found that antibody responses were dose-dependent, with 11 out of 12 stage I-III patients, and 8 out of 14 stage IV patients treated at 250 μg achieving a response [59]. Detailed clinical response data were not published from this study.
Ajani et al. [60], studied the addition of cisplatin (100 mg/m2 IV on day 1 of each 28 day cycle) and 5-FU (1000 mg/m2/day continuous IV infusion on days 1–5) to G17DT in 103 patients with untreated metastatic or unresectable gastric or gastroesophageal adenocarcinoma as part of an international phase II study. Ninety six patients were evaluable (seven patients were overdosed on 5-FU and were not analysed), and only 79 were evaluable for response (24 patients were excluded; one patient did not receive therapy according to protocol, and 23 patients did not undergo follow-up assessment). Of these patients, there was a 50 % best overall response rate (ORR), but the confirmed ORR was only 30 %. In the intent-to-treat (ITT) population, best ORR was 42 % and confirmed ORR was 25 %. mOS was 9.0 months and was significantly longer in the 90 % of patients who had a positive antigastrin antibody titre by ELISA than the 10 % who did not (10.3 months vs. 3.8 months; P < 0.0001) [60]. Grade 3–4 AE were notable for neutropenia (24 %), leukopenia (9 %), anaemia (12 %), febrile neutropenia (7 %), thrombocytopenia (5 %), nausea (20 %), vomiting (20 %), stomatitis (19 %), diarrhoea (5 %), myositis (13 %), fatigue (12 %), anorexia (12 %), dehydration (11 %), peripheral neuropathy (9 %), and syncope (5 %). Survival statistics were comparable with published data on 5-FU/cisplatin regimens, although there was a possible added benefit for immune responders in this trial.
In another phase II study, Gilliam and colleagues [61] randomised 154 patients with advanced pancreatic cancer who were unwilling or unable to undergo chemotherapy to receive G17DT or placebo. mOS was longer in patients who received G17DT (151 days vs. 82 days, P = 0.03), and those patients who developed antibody responses (73.8 % of the G17DT arm) also had longer mOS than those who did not respond, or those taking placebo (176 days vs. 63 days vs. 83 days, respectively; log-rank test, P = 0.003). However, in the ITT population, mOS was not statistically improved following G17DT administration (150 days vs. 83 days with placebo, P = 0.054). To summarise, although well tolerated, and with apparently improved survival in immune responders, G17DT effected only a trend toward significantly improved mOS in this population overall.
Finally, a phase II study by Rocha-Lima et al. [62], treated 161 patients with mCRC who had progressed on an irinotecan-containing regimen with G17DT plus irinotecan. In the ITT population, there were very few clinical responses (3 % had partial responses (PRs), 32 % had stable disease (SD)). Sixty-two percentage of patients had an immune response, and these patients had improved mOS compared with non-responders (9.0 vs. 5.6 months, P < 0.001) [62].
G17DT showed only modest antitumour efficacy in early phase clinical trials and has thus not been further pursued.
CD55
CD55, decay accelerating factor is a complement regulatory protein overexpressed by cancer cells to prevent them from complement-mediated destruction. 105 AD7 is a human anti-idiotype monoclonal antibody that mimics CD55 and stimulates CD4 and CD8 antitumour responses in patients with appropriate haplotypes (HLA/A1,3,24 and HLA/DR1,3,7) [63]. Interestingly, CD55 is expressed on 70–80 % of CRCs [64, 65], and a small neoadjuvant study treated 35 patients with resectable CRC with 105 AD7 before undergoing surgery. Approximately 83 % of patients expressing a permissive haplotype had antitumour responses, whereas only 12 % of patients without these haplotypes had a response. Sixty-five percentage of patients were disease-free at a median follow-up time of 4 years [66]. Maxwell-Armstrong and colleagues [67] randomised 162 patients with advanced CRC to 105 AD7 or placebo. Only 50 % of patients received the 3 planned doses of vaccine, and mOS was not prolonged with 105 AD7 (124 days vs. 184 days with placebo, P = 0.38). Finally, Ullenhag et al. [68], randomised 67 patients with resectable CRC 2:1 to neoadjuvant/adjuvant 105 AD7 (with or without BCG) or no treatment. Fifty-three percentage of immunised patients had an immune response on ELISPOT or proliferation assays, but survival was not assessed in this study. Therefore, despite demonstration of immunogenicity, 105 AD7 has not yet been shown to have a survival benefit in CRC.
CD17-1A
CD17-1A (edrecolomab) is an anti-idiotype murine monoclonal antibody that targets the tumour-associated glycoprotein GA733-2 [69]. Early clinical studies using CD17-1A monotherapy in advanced gastrointestinal cancers, and CD17-1A in combination with 5-FU, adriamycin, and mitomycin (FAM) in patients with advanced pancreatic cancer demonstrated that CD17-1A is well tolerated [70, 71]. Two other studies combined CD17-1A with interferon gamma in mCRC and advanced pancreatic cancer but failed to show a clinical benefit or a clear correlation between antibody development and improved survival [72, 73]. A larger phase II trial of CD17-1A monotherapy in advanced pancreatic cancer patients also failed to demonstrate efficacy [74].
Despite these findings, CD17-1A was tested in a large adjuvant CRC trial. A total of 189 patients with fully resected Dukes’ stage C CRC were randomised to adjuvant CD17-1A or observation. The overall death rate was decreased by 32 % in the CD17-1A arm compared with no treatment (log-rank P = 0.01), and the recurrence rate was decreased by 23 % (log-rank P = 0.07) [75, 76]. Also, the incidence of distant metastases was significantly reduced in the CD17-1A arm (log-rank P = 0.004) but local recurrences were not (log-rank P = 0.83) [76]. This study demonstrated that CD17-1A might be of clinical benefit in the adjuvant setting.
The addition of granulocyte-macrophage colony-stimulating factor (GM-CSF) to augment antibody-dependent cellular cytotoxicity (ADCC) conferred clinical benefit in a study of 20 patients with mCRC treated with CD17-1A: 2 patients achieved a complete response (CR), one achieved a partial response (PR), and 3 experienced stable disease (SD) for more than 3 months [77]. In another study of 20 patients with mCRC, this protocol was extended to include the addition of IL-2 to CD17-1A and GM-CSF. One patient on this treatment experienced a PR and 2 had SD [78]. Thus, IL-2 did not appear to improve the clinical efficacy of this regimen.
Finally, CD17-1A was studied in a phase III trial as adjuvant therapy in stage II colon cancer [79]. A total of 377 patients were randomised 1:1 to CD17-1A or observation, and the study was terminated early due to discontinuation of drug supply. Neither mOS nor disease-free survival (DFS) were significantly different between the study groups [79]. Similarly, a study of CD17-1A as adjuvant therapy in 2761 patients with stage III colon cancer randomised patients 1:1:1 to receive 5-FU/leucovorin with CD17-1A, 5-FU/leucovorin alone, or CD17-1A alone. There was no difference in 3-year OS between combination treatment and chemotherapy alone (74.7 % vs. 76.1 %; P = 0.53). Additionally, DFS was significantly reduced following CD17-1A treatment alone compared with chemotherapy alone (53.0 % vs. 65.5 %; P < 0.0001) [80]. Although well tolerated, the addition of CD17-1A did not show any clinical benefit.
To summarise, targeting CD17-1A has yet to show promise in large clinical trials despite early signals of potential benefit in the adjuvant setting for CRC.
GV1001
Telomerase, expressed by 85–90 % of human cancer cells while restricted in normal cells, functions to preserve telomeres at the end of chromosomes, thus promoting immortalisation. GV1001 is a telomerase peptide that therefore specifically targets malignant cells [81, 82]. In a phase I/II study of 48 patients with unresectable pancreatic cancer, 24 of 38 evaluable patients had immune responses, and these immune responders had significantly improved mOS (216 days vs. 88 days, P = 0.0001) [82]. Patients in the intermediate dose group had the longest mOS (260 days) compared with the low dose (119 days, P = 0.006) and high dose groups (153 days, P = 0.05).
A phase II trial of a combination of GV1001, GM-CSF, and cyclophosphamide in 40 patients with advanced hepatocellular carcinoma (HCC) did not demonstrate any objective tumour responses, and only 46 % of patients had SD on this regimen [83].
Finally, a large phase III trial (the TeloVac trial) focused on the treatment of locally advanced or metastatic pancreatic cancer patients with gemcitabine and capecitabine, with or without GV1001/GM-CSF. This trial failed to show an overall survival benefit following the addition of GV1001 to standard chemotherapy [84]. Patients (n = 1062) were randomised 1:1:1 to receive chemotherapy alone, sequential chemotherapy and GV1001/GM-CSF, or concurrent chemotherapy and GV1001/GM-CSF. mOS was not significantly different in the chemotherapy group when compared with the sequential group (7.9 months vs. 6.9 months, HR 1.19, 98.25 % CI 0.97–1.48, P = 0.05) or the concurrent group (8.4 months, HR 1.05, 98.25 % CI 0.85–1.29, P = 0.64) [84].
WT1
Wilms tumour gene (WT1) peptide-based cancer vaccines have been studied in pancreatic and biliary tract cancers. In a phase I study, 32 patients with advanced pancreatic cancer received a WT1 vaccine in combination with gemcitabine [85]. Within the 29 patients who completed 2 cycles of treatment, notable grade 3–4 AEs included neutropenia (44.8 %), leukocytopenia (27.6 %), lymphopenia (27.6 %), anaemia (6.9 %), and non-neutropenic biliary infection (24.1 %). There was one episode of cerebrovascular ischemia, a dose-limiting toxicity. Otherwise, AEs were comparable to those observed in patients receiving gemcitabine alone. Fifty-eight percentage of patients on study developed a delayed-type hypersensitivity (DTH) reaction, which was indicative of an immune response, and responders had improved mOS (10.9 months vs. 3.9 months, log-rank P = 0.003) [85]. A similar phase I study of WT1 peptide vaccine plus gemcitabine in 25 patients with advanced pancreatic or biliary tract cancers was disappointing. Although 59 % of patients had an immune response, this unfortunately did not translate into clinical benefit (mOS was 288 days for biliary cancer patients and 259 days for pancreatic cancer patients) [86]. WT1 vaccines have been extensively studied in a number of cancers, but at this point there are no published phase II or III data for WT1 vaccines in gastrointestinal cancers.
DNA Vaccines
Plasmid DNA Targeting CEA
Plasmid DNA vaccines targeting CEA have been investigated with limited success. In a phase I trial, Staff et al. treated patients with adjuvant CRC with a plasmid DNA vaccine construct containing a modified CEA gene fused to a T helper epitope of tetanus toxoid (CEA 66 DNA) [87]. GM-CSF was also administered, as was cyclophosphamide (to deplete regulatory T-cells). Ten patients received this regimen and only 2 had recurrent disease. The regimen was well tolerated with no evidence of grade 3 or 4 AEs.
Conry and colleagues studied a DNA vaccine encoding both CEA and hepatitis B surface antigen in 17 patients with mCRC [88]. Unfortunately only five patients had SD and none of the patients developed CEA-specific antibodies.
CEA DNA vaccines have not been further explored in clinical trials.
Whole Tumour Cell Vaccines
GVAX/CRS-207
GVAX is a lethally-irradiated allogeneic GM-CSF-secreting whole cell pancreatic tumour vaccine that has been studied alone and in combination with CRS-207, a live-attenuated Listeria-based vaccine expressing mesothelin (a cell surface tumour-associated antigen expressed on the majority of pancreatic cancer cells and thought to be implicated in cell adhesion and metastasis) [89, 90].
In a phase I study, Jaffe et al. first demonstrated safety and immune activation in 14 pancreatic cancer patients treated with GVAX in the adjuvant setting, delivered before and after chemoradiation [91]. A single-arm phase II study of 60 patients in the same setting showed a median DFS of 17.3 months (95 % CI 14.6–22.8 months) and mOS of 24.8 months (95 % CI 21.2–31.6 months) [92]. Treatment was well tolerated; the only grade 3–4 AE was eosinophilia, which was observed in two patients.
Cyclophosphamide was added 1 day before GVAX treatments in order to inhibit regulatory T-cells and enhance the anti-tumour immune response. Laheru et al. [93], enrolled 30 patients treated with GVAX alone, and another 20 patients treated with pre-vaccine cyclophosphamide. Although not randomised, patients who received GVAX alone had a mOS of only 69 days, as opposed to 130 days for patients who received cyclophosphamide prior to GVAX administration. Encouragingly, treatment was also relatively well tolerated. To investigate the addition of CRS-207, Le and colleagues [94] randomised 93 patients with pre-treated metastatic pancreatic cancer 2:1 to receive a combination of cyclophosphamide and GVAX or cyclophosphamide, GVAX, and CRS-207. mOS was improved in the CRS-207 arm in both the full analysis (6.1 months vs. 3.9 months; P = 0.0343) and per-protocol analysis (9.7 months vs. 4.6 months; P = 0.0335). The treatment was well tolerated; the only grade 4 AE in the CRS-207 arm were lymphopenia (two patients). This was the first study to establish a survival advantage using immunotherapy in pancreatic cancer.
Next, investigators attempted to augment this immune response using checkpoint inhibitors. The anti-cytotoxic T lymphocyte-associated antigen-4 (anti-CTLA-4) antibody—ipilimumab—was combined with GVAX in a phase Ib study of 30 patients with previously treated advanced pancreatic cancer [95]. Patients were randomised 1:1 to receive 10 mg/kg IV ipilimumab alone or in combination with GVAX, administered every 3 weeks for 4 doses followed by maintenance dosing every 12 weeks. mOS was improved in the combination arm, but unfortunately, this finding was not statistically significant (5.7 months vs. 3.6 months; P = 0.072) [95]. AEs were comparable between the study groups (grade 3–4 events included one episode of colitis in each arm and one episode of pneumonitis in the combination arm).
An ongoing phase II trial (STELLAR) is investigating the addition of nivolumab, a humanised IgG4 anti-programmed cell death protein 1 (anti-PD-1) antibody, to GVAX/CRS-207 and cyclophosphamide in patients with previously treated metastatic pancreatic cancer (NCT02243371). An estimated 108 patients will be randomised 1:1 to receive cyclophosphamide and GVAX/CRS-207 with or without nivolumab (3 mg/kg IV every 2 weeks), we await the results of this trial with interest. [96]
OncoVAX
OncoVAX is a vaccine comprised of autologous tumour cells with the immunomodulating adjuvant BCG. A phase III trial of OncoVAX in stage II and III colon cancer patients in the adjuvant setting randomised 254 patients 1:1 to receive OncoVAX or no further treatment [97]. In the ITT population, there was a trend toward OS benefit in stage II patients treated with OncoVAX (5-year OS: 82.5 % vs. 72.7 %; HR 0.544, 95 % CI 0.276–1.071; log-rank P = 0.074) but not among stage III patients (63.6 % vs. 70 %; HR 1.168, 95 % CI 0.522–2.469; log-rank P = 0.685) [97]. Given the lack of statistically significant survival benefit, OncoVAX has not been further studied in the stage II and III colon cancer setting.
Algenpantucel-L
Algenpantucel-L is an irradiated whole-cell allogeneic pancreatic cancer vaccine composed of two human pancreatic adenocarcinoma cell lines (HAPa-1 and HAPa-2) with a retrovirally inserted murine α(1,3)-galactosyltransferase (αGT) gene [98]. Expression of the αGT enzyme causes hyperacute rejection with complement and antibody-dependent cytotoxicity [98–102]. In a single-arm phase II study, 73 patients with resected pancreatic cancer received adjuvant algenpantucel-L, gemcitabine, and chemoradiation with infusional 5-FU. Median DFS was 14.1 months, and the treatment was well tolerated with no grade 4 adverse events attributed to algenpantucel-L. This survival outcome compared favourably to historical controls (11.4 month median disease-free survival in the RTOG-9704 trial; adjuvant 5-FU-based chemoradiation plus gemcitabine) [103]. The phase III PILLAR trial of algenpantucel-L in locally advanced pancreatic cancer has completed enrollment of 302 patients, randomised 1:1 to FOLFIRINOX plus algenpantucel-L or FOLFIRINOX alone; however, results have not yet been published (NCT01836432).
Vector-Based Vaccines
A vector-based vaccine approach can take various approaches to DC activation. These agents have in common the expression of, or encoding of, the target antigens. The use of a foreign particle provides a method to initiate the innate immune response, which can then stimulate the adaptive immune response against the specific target, as discussed in specific cases below. There are advantages and disadvantages to each vector, which have to be carefully considered during development of a vaccine and decisions on how it will be used clinically. Specifically, if the vector requires infectious capability to be efficacious, host-neutralising immunity against the vector can be a major limitation. Similarly, underlying hypersensitivity to a vector may also be a limitation.
Yeast Vectors
A yeast platform provides an example of a vector that does not initiate DC infection to trigger activation. Instead, this strategy exploits the underlying innate immunity against yeast particles to drive DC and then T cell activation. The transgene for the target antigen is transfected into yeast; the yeast cells are then grown in culture until they express the target antigen, after which they are heat-killed. This results in a non-infectious delivery mechanism for the target antigen that will be engulfed by DCs and trigger signalling through TLRs in a pathogen-associated molecular pattern (PAMP) [104]. The activated DCs then produce type I cytokines, including IL-12 and TNF-α, and induce T cell activation against the antigens they are presenting, which include the tumour-associated antigen (TAA) target of the vaccine. In preclinical models, yeast-based vaccines have been seen to induce activated T cells [105] that are capable of killing tumour cells expressing the TAA in vitro, which was found to slow tumour growth and improve OS in murine models [106].
Multiple clinical trials of yeast-based vaccines demonstrated the safety of the platform and the ability to generate specific T cell responses against a variety of target antigens, including KRAS (GI-4000), CEA (GI-6207), and brachyury [107–109], a transcription factor involved in the epithelial to mesenchymal transition in many adenocarcinomas [110–112]. The RAS, CEA, and brachyury targets are all potentially applicable in GI malignancies. In a phase 1 clinical trial of the yeast-brachyury vaccine, 17 of 31 evaluable patients developed brachyury-specific CD4 and/or CD8 T cell responses post-vaccination, and the proportion of responders increased at the higher dose levels (5 of 6, 83 %, at the highest dose level) [113]. These immune responses were observed in the context of minimal AEs, most commonly injection site reactions; no dose-limiting toxicities (DLTs) were observed. Clinical efficacy of these agents, as with most vaccine platforms, is an area of ongoing investigation.
Viral Vectors
A commonly employed platform for therapeutic cancer vaccines is a genetically modified virus. These viral vectors typically have been genetically modified to reduce the infection risk and immunogenicity of the viral particle to allow for multiple safe administrations, and increase the likelihood of a targeted T cell response against the target antigen. Modified viral vectors are commonly altered to include the transgene for the target protein, resulting in translation of that transgene into expression of the target protein within the infected cell. In most cases, the target of infection is a subcutaneous DC. Once infected, the DC will translate the full-length protein and then present it via MHC. Some viral vectors also include mechanisms to help activate the DC, triggering migration to the draining lymph node and presentation of the target antigen in combination with costimulatory molecules, providing signal 1 and signal 2 to drive T cell activation against the target antigen [114].
Poxviral Vectors
The poxviral platform has a long history in development as therapeutic cancer vaccines. Initial studies were performed using recombinant vaccinia (rV) across a variety of target antigens. rV-CEA was the first vaccinia vector tested in humans, including patients with GI malignancies. Evaluation of the rV platform demonstrated that host neutralising immunity of the vaccinia vector limited its applicability for booster dosing [115]. Hodge et al. later demonstrated that priming with vaccinia followed by booster dosing with replication-incompetent poxviral vectors, including canarypox and fowlpox, allowed for administration of multiple booster doses, resulting in greater T cell activation and persistence in preclinical models [116]. This prime and boost strategy proved to be effective in a clinical trial conducted by Marshall and colleagues using rV-CEA followed by avipox-CEA boosts [117]. Present strategies include further refinement of the vaccinia priming dose to minimise safety risk. To that end, a modified version of vaccinia, called MVA (modified vaccinia Ankara) has become the priming dose of choice in studies of poxviral vaccines [118].
Related posts:
 PD1 and PD-L1 Immune Checkpoint Inhibitors in Gastrointestinal Cancer
PD1 and PD-L1 Immune Checkpoint Inhibitors in Gastrointestinal Cancer
 Hypermutated Colorectal Cancer and Neoantigen Load
Hypermutated Colorectal Cancer and Neoantigen Load
 The Role of the JAK/STAT Signalling Pathway in Immunoregulation of Gastrointestinal Cancers
The Role of the JAK/STAT Signalling Pathway in Immunoregulation of Gastrointestinal Cancers
 Cancer Immunology
Cancer Immunology
 Endocrine Malignancies
Endocrine Malignancies
 PGE2-Associated Inflammation and Gastrointestinal Tumorigenesis
PGE2-Associated Inflammation and Gastrointestinal Tumorigenesis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


