2The Willink Biochemical Genetics Unit, Genetic Medicine, St Mary’s Hospital, Manchester, UK
Fabry disease (Online Mendelian Inheritance in Man #301500) is a rare X-linked metabolic disorder caused by the partial or complete deficiency of the lysosomal enzyme, α-galactosidase A.
Epidemiology
Fabry disease is generally considered to be the second most prevalent lysosomal storage disorder, after Gaucher disease, with an estimated incidence ranging between 1:40,000 and 1:170,000 persons. It may be much more common, and a recent study showed that 1:3,200 neonates in northern Italy have missense mutations in the alpha-galactosidase A gene, but many of these may be clinically silent. The diagnosis is often missed in childhood. All ethnic groups are affected. Although it is sex linked, heterozygous females are frequently symptomatic. The mechanisms underlying this are unknown, but may relate to skewed X inactivation. Up to 3–4% of adult male patients with cryptogenic stroke or unexplained left ventricular hypertrophy may have atypical Fabry disease.
Genetic basis
The AGAL A (GLA) gene is located at Xq22.1 and approximately 600 mutations have been identified (mainly missense mutations but also nonsense mutations and single amino acid deletions and insertions). Most of these mutations are ‘private’, having been identified only in individual families, while others, located at CpG dinucleotides (e.g. R227X), have been found as independent mutational events.
Pathophysiology
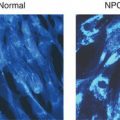
Neutral sphingolipids with terminal α-galactosyl residues (predominantly globotriaosylceramide (Gb3)) accumulate in the lysosomes of cells in a range of different organs and tissues. This includes endothelial, perithelial and smooth muscle cells of blood vessels, epithelial cells of glomeruli and tubules of the kidneys, cardiac myocytes, ganglion cells of the autonomic system, cornea and histiocytic and reticular cells of connective tissue. An accumulation of lysosphingolipids (i.e. glycosphingolipids that do not have N-acylated fatty acids) may be of particular significance in Fabry diseases and may lead to biochemical and structural changes which damage the cells. Abnormal proliferation of cells, e.g. of the intimal musculature of blood vessels, may be a part of the vascular pathology of Fabry disease.
Clinical presentation
Fabry disease is a progressive, multisystem disorder which typically results in a global reduction in quality of life of affected individuals (see Box 7.1). Symptoms in childhood typically relate to lethargy, tiredness, pain, cutaneous abnormalities (Figure 7.1), changes to sensory organs and often gastrointestinal disturbances. In early adulthood patients may suffer extension of any of the above symptoms and often develop lymphodema, proteinuria and the first signs of renal, cardiac or central nervous system/cerebrovascular disease. A characteristic facial appearance has been noted (Figure 7.2). In late adulthood (age >30 years) symptoms include worsening of the above and more severe organ dysfunction (cardiac disease, renal disease and cerebrovascular disease). The late onset forms of Fabry disease may present as stroke/TIA, left ventricular hypertrophy or renal failure.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


