Gaucher disease
Gaucher disease was first described by Philippe Gaucher in 1882. He was able to detect the abnormally enlarged cells in the spleen but he erroneously identified the condition as a primary neoplasm. In 1965, the primary defect was recognized as the inability of the mutated enzyme β-glucocerebrosidase to degrade glucocerebroside adequately. The activated macrophages, or “Gaucher” cells, harbour the accumulated undegraded substrate. Purification of the enzyme with subsequent cloning of the gene in 1985 led not only to identification of the many mutations that cause the disease but also to the concept of management. Enzyme replacement therapy (ERT) was successfully tested in a clinical trial in 1991 [1] leading to a revolution in the management of the disease. A decade later, substrate reduction therapy (SRT) was evaluated with potential to also impact the neurological manifestations of Gaucher disease [2].
Epidemiology
Gaucher disease is inherited as an autosomal recessive disorder. It is a panethnic disorder like the other lysosomal storage disorders but has a predilection among Ashkenazi Jews with a carrier frequency of 1: 17 and an expected birth frequency of 1: 850. Two distinct neuronopathic forms of Gaucher disease are common in Northern Sweden and in the area of the Palestinian town of Jenin, respectively. The estimated global frequency of Gaucher disease is 1: 50,000 to 1: 100,000.
Figure 6.1 Most common Gaucher disease mutations in the β-glucocerebrosidase gene. (Courtesy of Prof. Gregory A. Garbowski, MD, Cincinnati Children’s Hospital Medical Center.)
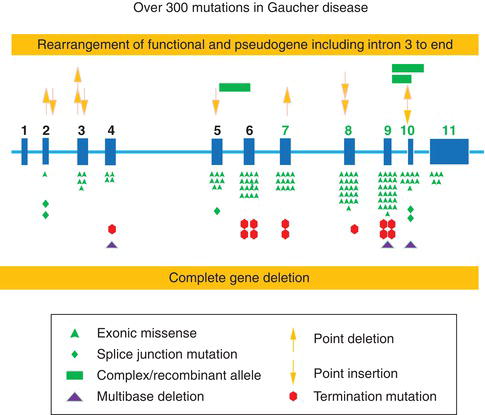
Etiology and pathogenesis: genetic basis
The glucocerebrosidase gene is located on chromosome 1q21. More than 300 mutations have been described (Figure 6.1) with an updated list published in 2008 [3]. The most common mutation, N370S, accounts for approximately 75% of the mutant alleles among Ashkenazi Jewish patients and approximately 30% among non-Jewish patients. Patients homozygous for N370S tend to have relatively mild phenotype; N370S is generally considered “protective” of development of neuronopathic features [4].
Mutations causing protein misfolding may induce endoplasmic reticulum (ER) associated degradation (ERAD) by the ER quality control system. In order to also avoid the proteotoxic effect of the mutated protein, and to circumvent ERAD, the new therapeutic modality of pharmacological chaperones (PC) [5] has been evaluated, facilitating trafficking from ER to Golgi to lysosome, and ultimately reducing storage of glucocerebroside in the lysosome.
Clinical forms
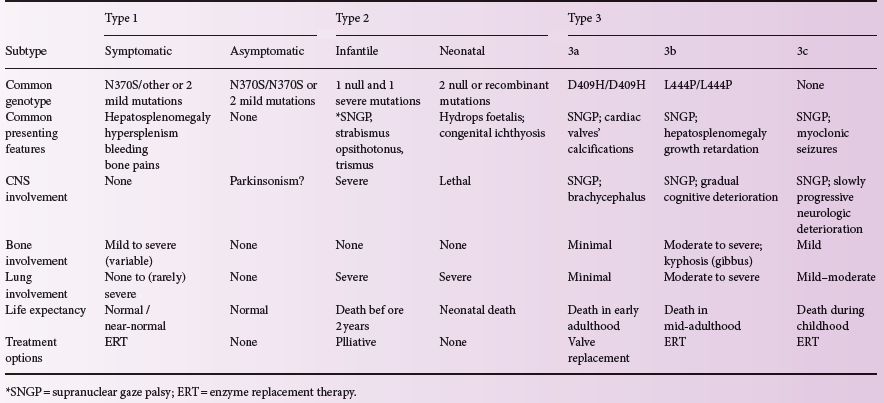
Three clinical forms have been defined, based on absence (type 1) or presence (types 2 and 3) of neurological signs.; Such classification is useful when talking about management options and genetic counselling.
Type 1
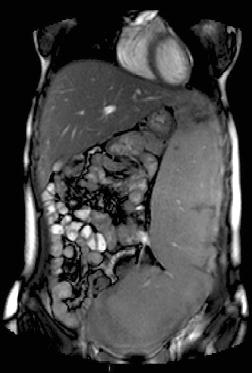
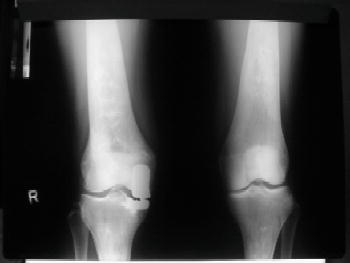
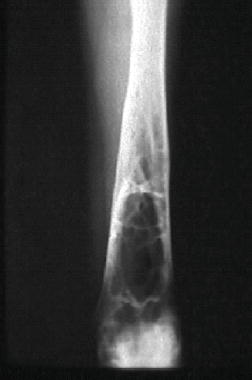
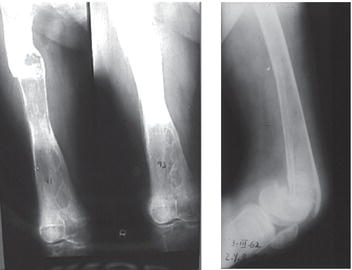
Type 1, the putatively non-neuronopathic form, is the most prevalent. The phenotypic spectrum ranges from virtually asymptomatic (these patients typically are diagnosed incidentally) to severe life-threatening disease. Age of onset of signs and symptoms and disease course are accordingly variable; the younger the age at presentation the more severe the clinical course. Anaemia and/or thrombocytopenia are common presenting findings, as is hepatosplenomegaly (Figure 6.2), which even in children may be massive. In children, too, height retardation may be noted [6] although there is catch-up growth in most cases irrespective of intervention. Erlenmeyer flask deformity of the distal femur (Figure 6.3), osteopenia (Figure 6.4), and varying degrees of bone pain are common.
Symptomatic skeletal features, including “bone crises”, osteonecrosis of joints, and pathological fractures of long bones (Figure 6.5) and vertebrae with collapse (Figure 6.6), although less prevalent, are, however, the leading causes of morbidity [6, 7]. Various imaging modalities assess/quantify bone involvement, but pre-symptomatic prediction of onset and rate of progression of bone disease has not yet been achieved. Lung involvement, especially infiltrative disease (i.e. of “Gaucher” cells), is an uncommon but serious complication usually in young children with severe genotype (including type 3) or in splenectomized adult patients with severe liver involvement. “Gaucher” cell infiltration can also be seen in other tissues and organs, notably the lymph nodes in various locations (Figure 6.7). Patients with type 1, despite being “non-neuronopathic”, may be at some increased risk for Parkinsonism and/or may develop peripheral neuropathies [6, 7].
Figure 6.2 Massive splenomegaly reaching into the true pelvis in a 20-year-old female with a slightly erratic history of imiglucerase therapy since age 4 years.
Type 2
Type 2 is a lethal neuronopathic form associated with homozygosity or compound heterozygosity for severe and/or null mutations and is characterized by hypertonic posturing, strabismus, trismus and retroflexion of the head shortly after birth; death following aspiration pneumonia and/or apnea/laryngospasm usually occurs by 2 years. Massive hepatosplenomegaly and lung involvement are common [6, 7].
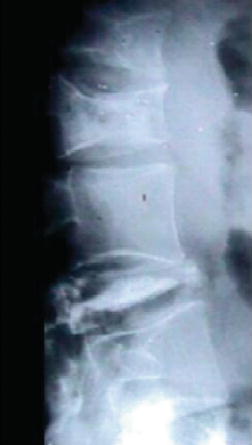
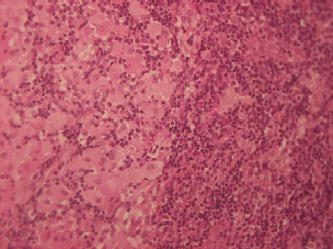
Figure 6.7 Biopsy of the parotid gland of a splenectomized 45-year-old male patient with a parotid gland adenoma alsoshowing “Gaucher” cell infiltration despite 18 years of treatment with imiglucerase.
Type 3
Type 3 is both a more heterogeneous and a more attenuated neuronopathic form [6, 7], presenting in childhood and characterized by the pathognomonic sign of supranuclear horizontal gaze palsy (SHGP). Most patients have at least one L444P mutation or other missense/null mutation. There is a subset of patients who are homozygous for the D409H mutation and develop progressive calcification of heart valves.
Diagnosis
The gold standard for diagnosing Gaucher disease is the detection of reduced β-glucocerebrosidase activity in peripheral blood cells compared to same-day normal controls. The recently-introduced methodology to assess the enzymatic activity on a dry blood spot could be used for large-scale screening (e.g. research purposes) but it is commonly associated with both false-positive and false-negative results and hence cannot replace the gold-standard assay [7]. The enzymatic diagnosis should be combined with the identification of mutations at the DNA level, and here too, in order to avoid pitfalls, it should be performed by complete gene sequencing. Although still commonly performed, bone marrow aspiration and biopsies for the identification of Gaucher cells is not the appropriate diagnostic test.
Biomarkers
Biomarkers add quality assurance to biochemical diagnoses, but are more importantly used to quantify dynamic changes in the clinical course over time, reflecting disease progression (usually in the untreated patient) or clinical improvement with specific therapy. Chitotriosidase activity, which is elevated hundreds-fold in Gaucher disease, has been used in the past decade as the preferred surrogate marker [8], but, because of a genetic deficiency of chitotriosidase in ~6% of the population, CCL18 (PARC) levels are additionally evaluated. There is an ongoing search for new biomarkers (such as MIP-1alfa, MIP-1beta and others) to better predict future complications.
Routine follow-up of patients
At minimum, monitoring disease expression and the effect of ERT include serial evaluation of haematological parameters and reduction in hepatosplenomegaly. Although ultrasonography may be recommended for repeat assessments as the least invasive and having no risk of radiation, especially in children, it is the least used because of issues of reliability and reproducibility and the importance of observer experience. Computed tomography (CT) was employed in the past, but today, magnetic resonance imaging (MRI), especially because of justifiable concerns about radiation with periodic assessments, is preferred. Similarly, scintography is rarely justified. The most sensitive tool for the follow-up of bone involvement is probably Quantitative Chemical Shift Imaging (QCSI) but is the least available.
Table 6.1 presents all the subtypes of Gaucher disease and their characteristics.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


