Primary Myelofibrosis
Mayo Clinic, Rochester, MN, USA
Introduction
Primary myelofibrosis (PMF) is listed under the World Health Organization (WHO) classification category of myeloproliferative neoplasms (MPNs). The operational subcategory of “BCR–ABL1-negative MPN” includes PMF, polycythemia vera (PV), and essential thrombocythemia (ET). All three disorders are characterized by stem cell–derived clonal myeloproliferation and the presence of somatic mutations involving JAK2 (in the majority of patients) and MPL or other somatic mutations (in the minority) (Table 26.1). The pathogenetic relevance of JAK2 or other mutations in MPN is poorly understood and currently under investigation. Regardless, clonal myeloproliferation in PMF is associated with putatively reactive bone marrow (BM) fibrosis, osteosclerosis, angiogenesis, extramedullary hematopoiesis (EMH), and abnormal cytokine expression.
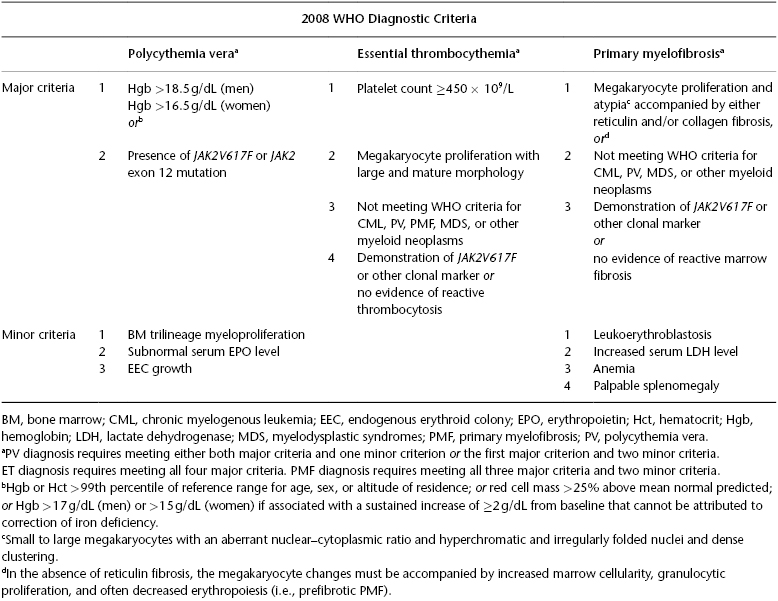
Table 26.1 World Health Organization (WHO) diagnostic criteria for polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PM) (Source: Tefferi A, et al. Blood. 2007;110:1092–7).
1. How is a diagnosis of PMF established?
PMF is diagnosed according to the WHO system. Post-PV or post-ET MF is diagnosed according to criteria set by the International Working Group for MPN Research and Treatment (IWG-MRT) (Barosi et al. 2008). Diagnosis of PMF is suspected in the presence of peripheral blood (PB) leukoerythroblastosis (LES; i.e., the presence of nucleated red cells, immature granulocytes, and dacryocytes). However, LES can result from any form of BM infiltration, including metastatic cancer or lymphoma. Therefore, a BM examination is always indicated in the presence of unexplained LES. BM morphology is key in diagnosing PMF and distinguishing it from reactive bone marrow fibrosis or other myeloid malignancies. In addition, the presence of JAK2V617F or of trisomy 9 and del(13q) is highly suggestive of the diagnosis.
2. What are the differences between the International Prognostic Scoring System (IPSS), dynamic IPSS (DIPPS), and DIPSS-plus?
Current prognostication in PMF is best accomplished by the use of the IPSS, DIPSS, or DIPSS-plus. The IPSS is applicable at the time of initial diagnosis and uses five independent predictors of inferior survival: age >65 years, hemoglobin <10 g/dL, leukocyte count >25 × 109/L, circulating blasts ≥1%, and the presence of constitutional symptoms. The presence of 0, 1, 2, and ≥3 adverse factors defines low-risk, intermediate-1 risk, intermediate-2 risk, and high-risk disease, respectively. The corresponding median survivals were 11.3, 7.9, 4, and 2.3 years. DIPSS and DIPSS-plus are applicable at any time during the disease course. DIPSS assigns two, instead of one, adverse points for hemoglobin <10 g/dL, and risk categorization is accordingly modified: low (0 adverse points), intermediate-1 (1 or 2 points), intermediate-2 (3 or 4 points), and high (5 or 6 points). The median survival was not reached in low-risk patients; it was 9.8 years in intermediate-1, 4.8 years in intermediate-2, and 2.3 years in high risk.
DIPSS-plus includes three additional DIPSS-independent risk factors: platelet count <100 × 109/L, red cell transfusion need, and unfavorable karyotype. The four DIPSS-plus risk categories based on the aforementioned eight risk factors (Table 26.2) are low (no risk factors), intermediate-1 (one risk factor), intermediate-2 (two or three risk factors), and high (four or more risk factors), with respective median survivals of 15.4, 6.5, 2.9, and 1.3 years. An unfavorable karyotype for the DIPSS-plus system includes a complex karyotype or sole or two abnormalities that include +8, −7/7q−, i(17q), inv(3), −5/5q−, 12p−, or 11q23 rearrangement. DIPSS-plus was recently further enhanced by the identification of patients with AML-like prognosis, defined by the presence of monosomal karyotype, inv(3) and i(17q) abnormalities, or any two of the following: circulating blasts >9%, leukocytes ≥40 × 109/L, or another unfavorable karyotype. Leukemic transformation in PMF is predicted by unfavorable karyotype and platelet count <100 × 109/L. Emerging data suggest an additional adverse prognostic effect from certain molecular markers (e.g., IDH, EZH2, SRSF2, or ASXL1 mutations) and increased serum interleukin 8 (IL-8) and IL-2 receptor levels or serum-free light chain levels. Interestingly, the additional prognostic value of somatic mutations was studied in 879 PMF patients and identified ASXL1 mutations as a DIPSS-plus independent risk factor. The combined consideration of CALR and ASXL1 mutations in PMF was also recently highlighted by showing the best survival in the presence of CALR and absence of ASXL1 mutation (i.e. CALR+ASXL1-) and worst survival in patients with CALR–ASXL1+ mutational status.
Table 26.2 Risk stratification and risk-adapted therapy in primary myelofibrosis (PMF)
| DIPSS-plusa risk groups PMF | Median survival | Management of PMF |
|---|---|---|
| Low risk (no risk factors)b | ∼15.4 years | Observation or conventional drugsc |
| Intermediate-1 risk (1 risk factor)b | ∼6.5 years | Observation or conventional drugsc or experimental drugs |
| Intermediate-2 risk (2 or 3 risk factors)b | ∼2.9 years | Allo-HCT or experimental drugs |
| High risk (≥4 risk factors)b | ∼1.3 years | Allo-HCT or experimental drugs |
DIPSS, Dynamic International Prognostic Scoring System.
aInformation in this column from Gangat N et al. J. Clin Oncol. 2011;9(4):392–7.
bDIPSS-plus uses eight risk factors for inferior survival: age >65 years, hemoglobin <10 g/dL, leukocyte count >25 × 109/L, circulating blasts ≥1% , presence of constitutional symptoms, presence of unfavorable karyotype, platelet count <100 × 109/L and presence of red cell transfusion need. Please note that a transfusion-dependent patient automatically has two risk factors because of transfusion need (one risk point) and hemoglobin <10 g/dL (one risk point).
cAndrogen preparations or thalidomide with prednisone for anemia; hydroxyurea for symptomatic splenomegaly.
3. Does diagnosis of PMF always warrant therapy?
Related posts:
 17: Management of Therapy-Related Myeloid Neoplasms
32: Hematopoietic Cell Transplantation in Chronic Lymphocytic Leukemia
59: Autologous Hematopoietic Cell Transplantation in Multiple Myeloma
17: Management of Therapy-Related Myeloid Neoplasms
32: Hematopoietic Cell Transplantation in Chronic Lymphocytic Leukemia
59: Autologous Hematopoietic Cell Transplantation in Multiple Myeloma
 11: Minimal Residual Disease in Acute Myeloid Leukemia
11: Minimal Residual Disease in Acute Myeloid Leukemia
 71: Secondary Brain and Spinal Cord Tumors
71: Secondary Brain and Spinal Cord Tumors
 130: Controversies Related to Oncology Clinical Trial Development
130: Controversies Related to Oncology Clinical Trial Development



