Introduction
The lysosomal storage disorders (LSDs) comprise a very heterogeneous group of diseases with regard to onset and diversity of symptoms. The heterogeneity and the low prevalence of individual LSDs can hamper early recognition and diagnosis. It is not uncommon that more than a decade passes before a proper diagnosis is finally made [1]. LSDs are generally progressive disorders and early diagnosis may be important for treatment decisions and genetic counselling. Therefore, screening in high-risk populations and carrier screening have been employed and newborn screening is the subject of intense debate. On the other hand newborn screening is already available in some countries. The most well known and earliest example of successful screening for LSDs comes from carrier screening for Tay–Sachs disease. In the early 1970s identification of carriers became feasible using enzymatic assays, later followed by mutation analysis. A joint program resulted in a dramatic decrease in the incidence of Tay–Sachs disease in the Ashkenazi Jewish population [2]. More recently, with the development of enzyme replacement therapy (ERT) and hematopoietic stem cell transplantation (HSCT), screening has focused on early identification of individuals who may benefit from timely intervention, i.e. before irreversible damage has occurred. For example, there is broad consensus that MPSIH (Hurler disease) patients should receive HSCT before the age of 2.5 years [3].
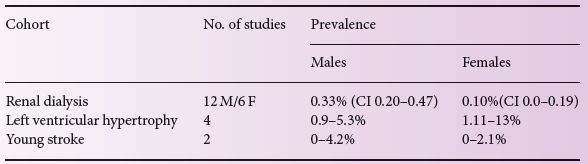
Table 23.1 Outcome of screening studies for Fabry disease in different cohorts.
However, for most LSDs the limited data on the natural course, especially of the attenuated forms, as well as the paucity of long-term follow-up data on the effectiveness of different treatments, hamper wide implementation of screening. Nevertheless, the developments and the technical advances in biochemical and genetic testing have resulted in several efforts towards expanded carrier, high risk and newborn screening.
Carrier screening
The purpose of carrier screening is to advise couples about the risk for recessive diseases in their offspring. In the Ashkenazi Jewish population, this has become an accepted way of preventing severe genetic diseases. Following this success, the approach has been expanded to other ethnic groups. Increasing knowledge on diversity in clinical expression may cause some concerns. For example, the Tay–Sachs heterozygote detection program at New York University Medical Center, New York, was expanded in January 1994 to include carrier testing for Gaucher disease. Since the most common form of Gaucher disease may exhibit very mild symptoms, carrier testing for this disorder does not necessarily prevent severe disease. Despite these concerns, the American College of Medical Genetics (ACMG) recently recommended carrier screening not only for Tay–Sachs and Gaucher disease but also for mucolipidosis type IV and Niemann–Pick disease type A in high risk groups [4]. While the lack of unequivocal genotype–phenotype relationships may hamper adequate predictions and counselling, the ethical discussion has fallen behind the advancement in genome sequencing programs and availability of commercial kits to screen young couples for many inherited disorders, including several LSDs.
High risk population screening
High-risk population screening involves the screening of patients with specific symptoms that could be caused by a LSD but where a LSD had not been considered. This has occurred particularly for Fabry disease after the introduction of ERT. Nakao et al. were the first to demonstrate an increased prevalence of Fabry disease (7/230, 3%) in males with left ventricular hypertrophy (LVH) [5]. This observation has led to additional screening studies in patients with unexplained LVH, but also in patients with end stage renal disease, young stroke and other symptomatic cohorts (summarized in Table 23.1, adapted from [6]). To our knowledge there are currently no systematic high-risk screening studies published for other lysosomal storage disorders.
Newborn screening
Early diagnosis of LSDs can avoid diagnostic odysseys and may result in timely initiation of treatment, which can be of great importance to patients and their families. For some disorders, early recognition of the disease and appropriate therapy can result in more favourable outcomes. This is particularly true for HSCT in some neurological LSDs such as Krabbe disease and MPS-1 H, although late complications can occur (see also case A). In infantile Pompe disease, early initiation of ERT is lifesaving, while irreversible skeletal complications may be prevented by early initiation of ERT in Gaucher disease. In Fabry disease, end organ damage is generally irreversible. These considerations form the rationale for the implementation of newborn screening programs. However, the long-term outcomes of HSCT and the effectiveness of pre-symptomatic initiation of ERT have not been fully elucidated. It is conceivable that therapeutic failure is perhaps too quickly ascribed to delayed intervention. Currently pilot programs for Fabry and Pompe disease are being employed in Taiwan and the state of Washington, USA. In some other US States Fabry, Pompe, Krabbe, Niemann–Pick A and Gaucher disease have been added to the newborn screening programs. Recently, the AMCG Working Group on diagnostic confirmation of LSDs has published an excellent educational guideline for the diagnostic approach and management of these LSDs [7].
Methodological issues of screening
A wide range of laboratory tests have been used to detect the presence of an LSD in a given population (be it newborn or a symptomatic cohort). Chamoles et al. [8] were the first to demonstrate that blood spots can be used to measure a variety of different lysosomal enzymes with artificial substrates. Other methods, which are still experimental, include multiplexing techniques to measure several lysosomal enzymes at once and the detection of accumulated substrate in blood or urine samples. Despite the tremendous technical progress, many challenges remain. For example, enzymatic analysis may fail to detect some individuals (e.g. females with Fabry disease) or may detect patients with a pseudodeficiency, who will never develop disease (e.g. arylsulfatase A deficiency without pathologic evidence of storage) [7]. Confirmation and interpretation by skilled biochemical laboratories is needed to avoid misdiagnosis. Undoubtedly, the most promising technique that will have a profound impact on screening is whole genome or next generation sequencing. While this technique is currently predominantly used to identify the genetic cause of well-defined phenotypes, the rapidly declining costs of whole genome sequencing may ultimately result in its use as a screening tool. While this technique may circumvent the aforementioned issues with false-negative or false-positive results regarding enzyme activity analysis, it may create uncertainty when (new) mutations with unknown clinical significance are identified (see case B).
Considerations of screening
In order for a disease to be included in a neonatal screening program, several criteria need to be met. While Wilson and Jungner’s criteria in 1968 (listed in Box 23.1) still form the basis of the decision to include diseases in a newborn screening program, these have been subject to intense debate. In 2006, the AMCG issued a report that newborn screening could be justified for family and societal benefits, even if there is no direct benefit for the screened infant. In countries outside the US, some disorders are included when adequate supportive care has been shown to have a profound effect on life expectancy and quality of life (e.g. cystic fibrosis). The same debate is ongoing for LSDs and will require input from patient groups and various specialties, including ethicists, biochemists and medical specialists.
One of the major concerns is the detection of late-onset variants or genetic variants of unknown clinical significance. As stated previously, the phenotypic spectrum of LSDs is broad and ranges from severe disease with early childhood mortality to late-onset disease with limited morbidity. Neonatal screening will identify patients from this entire spectrum. In order to distinguish those patients with LSDs who benefit from early treatment, a reliable prediction of the phenotype is mandatory. Genotyping can be helpful in some cases but in general genotype/phenotype correlation is poor in most LSDs [7]. Variants with uncertain clinical phenotypes will be identified and it is likely that the number of these uncertain or late-onset variants identified through screening will become the majority of the diagnosed subjects (as is clear from Table 23.2). An early diagnosis in a patient with unclear or late onset disease may cause anxiety, lead to societal problems such as higher premiums for health insurance and create loss of an unconcerned carefree life. Societal support for early detection of adult onset diseases is mixed [9] and has not been investigated for detection of abnormalities of uncertain significance. In addition to these concerns, much has to be learned about the long-term effectiveness of treatments, both for the classical as well as late-onset variants. Gradually, it has become clear that many LSDs are not cured by the currently available strategies. For example, pre-symptomatic bone marrow transplantation in Krabbe disease may not prevent motor disability, speech difficulties and growth failure [10]. Larger, collaborative studies are needed to explore these outcomes, and these need to be incorporated in the ethical discussions.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


