2 Department of Paediatrics, University of Padova, Padova, Italy
Introduction
Of the 50 or more described lysosomal storage diseases (LSDs) the majority are neuronopathic and associated with profound neurodegeneration. The blood–brain barrier (BBB) is created by the vascular bed of the central nervous system (CNS) and very closely regulates the composition of the brain extracellular fluid. The blood–brain barrier is a vital issue in neuronopathic LSDs as substrate reduction, chaperone and enzyme replacement therapies must obtain access to the CNS in order to treat the neurological aspects of the disease. It has been shown that the BBB is compromised in a number of the neuronopathic LSDs, especially those with an early onset. Dysfunction of the BBB and a compromise of its regulatory role in CNS homeostasis may contribute to the CNS pathology and neurodegeneration.
The LSDs are a unique model in which to study neurodegeneration. In fact, although the accumulation of intermediate degradation products is due to a single gene defect, this affects the whole cellular metabolic system causing neuronal death apoptosis or necrosis at advanced stages of the disease. CNS pathology causes mental retardation and progressive neurodegeneration that ultimately ends in an early death. Many of these features are also found in adult neurodegenerative disorders, such as Alzheimer, Parkinson or Huntington diseases. A lysosomal involvement in the pathogenesis of these disorders has been described. Improved knowledge of secondary events will have an impact on diagnosis, staging and the follow-up of affected children. Importantly, new insights may provide indications about possible advances in treatment and disease reversal.
CNS storage and neuropathology
A common characteristic of many LSDs is the onset and relentless progression of neurodegenerative signs and symptoms. This neurodegeneration may start early in life or not manifest until later, depending on the LSD and the nature of the enzyme deficit. The cascade of events that leads to neurodegeneration and the neuropathology is not simple and is certainly not related solely to the primary storage product. Many secondary storage products, such as gangliosides, accumulate and may be a principal cause of CNS damage. Abnormal inflammatory responses are also present in the brains of LSD patients and the microglia are “activated” and show abnormal activity and secretion of cytokines.
Many of the LSDs exhibit, as part of their CNS manifestation, increases in oxidative stress, alterations in calcium homeostasis, autophagy and altered lipid trafficking in addition to neuronal, glial and other CNS cell accumulation of the primary storage product. Primary and secondary events have a profound impact on the morphology and function of neural cells, producing alteration in dendritic processes with the generation of meganeurites and axonal spheroids together with somatic swelling. Meganeurites are huge axon hillock enlargements containing typical storage bodies, which often exceed the volume of adjacent perikarya. Meganeurites are probably associated with an increased expression of GM2-ganglioside, which probably acts as a neurite growth promoting factor or as an acceptor molecule for growth factors which influence the cells. Axonal spheroids are enlargements distal to the axon initial segment region. While meganeurites contain storage material consistent with the specific defective lysosomal hydrolase, and their morphology is therefore considered disease-specific, axonal spheroids contain a distinctly different array of materials with ultrastructural features (tubulovesicular profiles, mitochondria and dense and multivesicular-type bodies) that are the same across many types of storage disorders. While there is quite a clear understanding of possible mechanisms underlying the genesis of ectopic dendrites on cortical pyramidal neurons in LSDs, little is known regarding the functional consequences of this event. Asymmetrical synapses are known to occur on these dendrites and this input is likely to injure the neuron function. Evidence suggests that lysosomal storage impairs autophagy, and relies on the hypothesis that storage may interfere with lysosomal contribution to the autophagic process resulting in accumulation of polyubiquitinated proteins and dysfunctional mitochondria ultimately leading to apoptosis.
This becomes relevant since it has been hypothesized that treatment might be achieved by controlling autophagy. The lysosome is not an isolated “waste disposal” organelle within the cell, but is in communication with all the major cell domains. Mitochondrial abnormalities have been shown in many LSDs suggesting a common cause. As mitochondria are essential in the production of energy via ATP and lipid biosynthesis, alteration in their function causes activation of an ER stress response, mitochondrial apoptotic signalling, or both triggering cell death under physiologic or pathologic conditions. Depletion of ER calcium-stores or elevation of cytosolic calcium levels, can activate different biochemical pathways such as the cytoplasmic apoptotic pathway mediated by ER-localized caspase activity that in turn induces a form of ER stress, known as the “unfolded protein response” (UPR). The UPR system is aimed at minimizing protein production in the ER and increasing production of the chaperone proteins that can facilitate correct folding of the unfolded or misfolded proteins. It has been suggested that UPR may represent a common mediator of apoptosis in both neurodegenerative and non-neurodegenerative LSDs.
Calcium is a fundamental component of eukaryotic cells and plays a major role in cell homeostasis as an intracellular mediator. Excitable cells respond by alterations in cell signaling pathways, through calcium modulation. A high calcium concentration is toxic to neurons so at rest the cytosolic calcium concentration is maintained at low levels by the concerted action of specialized channels, a calcium pump, calcium-dependent enzymes, and calcium-binding proteins. These mechanisms are localized in the cytosol, ER and mitochondria, the three compartments that control the traffic of calcium across the plasma membrane or into intracellular stores. In neurons, calcium is stored mainly in the ER where it is pumped from the cytosol by the sarco/endoplasmic reticulum calcium ATPase (SERCA) and from which it is released back into the cytosol through two types of channel, one of which is a ryanodine receptor. Altered calcium homeostasis is one of the most common pathological features of lysosomal storage in LSDs. It has been described in several LSDs, including Gaucher disease.
Like calcium homeostasis, iron homeostasis is also essential in the CNS and furthermore it appears to play a role in the pathogenesis of neurodegenerative disease including Parkinson and Alzheimer diseases if altered. The exact mechanism by which iron causes this pathogenic consequence is still not clear. A recent study on murine models of GM1- and GM2-gangliosidoses, both of which are characterized by progressive depletion of iron in brain tissue, showed alterations in iron homeostasis. Progress in the understanding of such complex processes will open, together with a better comprehension of the development of the LSD phenotype, the application of new potential synergistic therapies to be coupled to the actual gold standard treatment, such as enzyme replacement therapy (ERT).
Normal function of the blood–brain barrier
The BBB enables the creation of an unique fluid environment for the brain which is extremely stable and much more tightly controlled than the extracellular fluid (ECF) of the soma. Regulation of the ionic composition of the brain ECF and cerebrospinal fluid (CSF) needs to be precisely controlled in order to allow the maintenance of stable resting potentials and the generation of reliable action potentials. The background concentrations of the multiple CNS neurotransmitters is also tightly regulated to reduce background noise at synapses in order to maintain the complex neural integrative processes that the CNS must perform. Also the total protein content of CSF is very low and is only 0.5% of that of plasma. The protein content of the CSF in foetal life is much higher and this promotes neurogenesis, gliogenesis and vasculogenesis. Some of these foetal proteins are transported into the brain and others are synthesized within the cerebral compartment. Once the brain is fully developed CSF proteins levels fall in early life in order to promote brain cellular structural stability, which is vital for correct and predictable CNS function.
A major role of the BBB is neuroprotection, as it protects the brain from blood-borne neurotoxins, which may be endogenous metabolites or acquired from the environment (Figure 21.1). The BBB is essentially formed by the endothelial cells of the cerebral capillaries which are the major exchange surface area (up to 20 m2 in the human) between the blood and the brain. The endothelial cells also form tight junctions between the individual cells of the capillary wall which are robust enough to prevent even the diffusion of small ions through the paracellular pathway and can create an electrical resistance in vivo of at least 2,000 Ω.cm2 across the endothelium. The fact that simple aqueous paracellular diffusion of solutes is prevented by the tight junctions means that all solutes entering or leaving the brain must cross the cerebral endothelium by a transcellular route.
Thus physicochemical interactions with the plasma membrane and glycocalyx of the endothelial cells and the presence of transport systems embedded in the membranes of endothelial cells facing the blood and brain will determine the permeability properties of the BBB. The blood–brain barrier possesses a spectrum of solute carriers (SLCs) which transport essential small molecular weight nutrients into the brain and several members of the ATP binding cassette (ABC) transporters, principally P-glycoprotein (PgP/MDR), breast cancer-related protein (BCRP) and a number of the multidrug resistance proteins (MRPs). These ABC transporters are energy-dependent and can transport solutes out of the brain against a concentration gradient.
Therapeutic delivery across the blood–brain barrier
The presence of tight junctions in the BBB between the endothelial cells will prevent the paracellular diffusion, via an aqueous pathway, of virtually any hydrophilic solute irrespective of molecular size. Table 21.1 summarizes the mechanisms by which therapeutics may cross the blood–brain barrier.
Small molecular weight entities, groups A to D, may show a variety of interactions with the BBB. Molecules that have a significant polar surface area and are not lipophilic (Group A) are generally excluded from the brain as, there is no paracellular pathway. Further, they are unable to dissolve in the lipid of the cell membrane from the water phase because of the Gibbs free energy required and consequently cannot cross the BBB by simple diffusion. These exclusion rules also apply to many polar metabolites that the brain requires and thus the substrate-specific solute carriers (SLCs) are embedded in the cell membranes of the endothelial cells that will transport essential metabolites across the barrier. These SLC transporters may simply facilitate diffusion or may be exchangers or be secondarily active in that they are powered by an ion-gradient. Importantly the expression of the carrier proteins can be different in the blood-facing and brain-facing membranes of the endothelial cells so that transport for specific substrates has a polarity either into or out of the brain. Furthermore, some small molecular weight therapeutics are sufficiently similar to an endogenous metabolite as to be recognized by these transporters and are carried into the brain (Group B). Generally this transport of drugs has been serendipitously discovered, for example with L-dopa, and only occasionally has been deliberately designed into the molecule as is the case with gabapentin. Molecules which are more lipid-soluble at physiological pH (Group C) can intercalate with the cell membrane and passively cross the BBB by simple diffusion. For these passively diffusing molecules, greater lipid solubility tends to enhance the rate of their brain penetration. However, a number of the more lipid soluble molecules are substrates for the ABC transporters and are actively ejected from the cerebral endothelium with little consequent brain penetration (Group D). Lipid-soluble molecules, which have a larger molecular weight (more than 8 nitrogen or oxygen atoms in their structure), tend to be acidic, have a more complex planar or branched structure, are likely to be substrates for the P-glycoprotein and breast cancer-related protein which are probably the major ABC effluxers in all mammals. The Multidrug Resistance Proteins (MRPs) are a futher group of ABC transporters. The substrates of ABC transporters may actually be removed from the cell membrane before they fully enter the endothelial cell. The degree to which molecules disturb the structure of the cell membrane, and the time they spend in the cell membrane (dwell-time), may relate to their likelihood of being a substrate for an ABC transporter and their chance of being recognized by the transporters. The MRPs tend to prefer substrates that have already undergone Phase 1 metabolism and are conjugated, for example sulfated or glucuronidated. This renders them less membrane-permeant and they are simply pumped out of the endothelial cells by the MRPs. The currently used substrate reduction therapeutics (SRTs) and chaperones all tend to be low molecular weight substances and their brain penetrance will be subject to the above considerations.
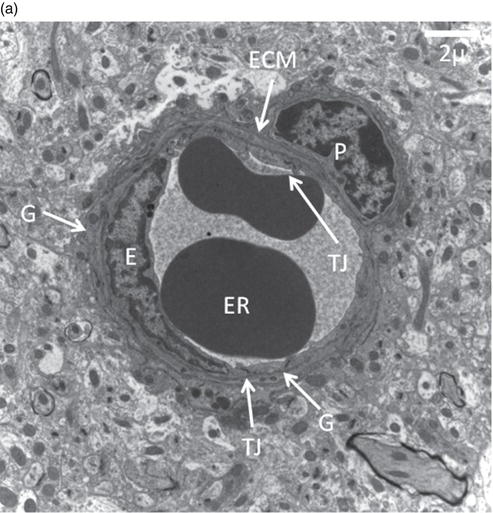
Figure 21.1 Blood–brain barrier in mouse.
(a) Cerebral capillary, site of the blood–brain barrier, in a normal adult C57B6 mouse containing two erythrocytes (ER). E,endothelial cell, with nucleus clearly visible; TJ, tight junctional complexes; P, pericyte; ECM, extracellular matrix surrounding thecapillary which has a different composition from that of the parenchymal matrix. The pericytes are responsible for secreting andre-modelling this perivascular extracellar matrix of which heparan sulfate and dermatan sulfate are major components; G, glial end feet.(Courtesy of Dr Anja Zensi.)
(b) Cerebral capillary from a symptomatic 8- to 10-month-old MPS IIIA mutant mouse showing significant pathology. *Large inclusion bodies in endothelial cell and the pericyte are probably late endosomes/lysosomes, loaded with the major storage product, heparan sulphate. The profile of the capillary is partially collapsed and the lumen is constricted. The borders of the extracellular matrix appear corrugated compared to the wild-type mouse. The cytoplasm of the endothelial cell is thickened and will present a greater diffusional barrier than in the normal mouse. The tight junctional complex appears normal and can be shown experimentally to effectively close the paracellular pathway to sucrose (MW 342.3), even at this late stage of the disease. (Courtesy of Dr Larisa Mihoreanu.)
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


