2 Professor of Medicine, University of Cambridge, Addenbrooke’s Hospital, Cambridge, UK
Introduction
This chapter includes sections on two typical storage disorders that have not been covered in other chapters – Farber disease and acid lipase deficiency – and a possible new lysosomal nucleoside transporter defect. There is also a discussion of the function and pathology of lysosomal proteases, the cathepsins, deficiencies of which lead to a wide range of phenotypes. Sections on disorders of biogenesis of lysosome-related organelles and the possible contribution of defects in the formation of the lysosomal recognition marker, mannose-6-phosphate, to non-syndromic stuttering, provide further evidence for the wide role of lysosomes.
Farber disease: acid ceramidase deficiency (OMIM: 228000)
Farber disease is a rare (less than 100 cases reported), autosomal recessive, lysosomal sphingolipid storage disorder caused by a deficiency of acid ceramidase (N-acylsphingosine amidohydrolase, ASAH1: EC 3.5.1.23). The genetically distinct alkaline ceramidase activity present in most cells is not affected in this disorder. The deficiency of acid ceramidase (OMIM: 228000) leads to the intralysosomal accumulation of ceramide in most tissues, including heart, liver, lung and spleen. Farber disease is also called Farber lipogranulomatosis because of the formation of the subcutaneous nodules near joints and other pressure points. Despite the small number of patients, Farber disease has been classified into seven subtypes according to age of onset, severity and which tissues are affected by the accumulation of ceramide. Types 1–5 are called classic, intermediate, mild, neonatal and neurologic progression, respectively. Type 6 arises from a serendipitous combination of Farber and Sandhoff diseases. In Type 7 a defect in prosaposin leads to a functional deficiency of acid ceramidase together with deficiencies of β-galactocerebrosidase and β-glucocerebrosidase activities. These patients present neonatally with a rapidly progressing neurovisceral lipid storage disease. Acid ceramidase is activated by saposin D in vitro but no patients with an isolated deficiency of saposin D have been reported. A mouse with a mutation in the saposin D domain of the prosaposin gene accumulated ceramide in the kidney and brain suggesting a role for saposin D in sphingolipid metabolism in vivo.
The characteristic features of Farber disease include progressive hoarseness due to laryngeal involvement, painful swollen joints, subcutaneous nodules and pulmonary infiltrations. Initial signs appear between 2 and 4 months of age except in the mild type 3. Death usually occurs within the first years of life but prolonged survival is known in patients without severe CNS involvement. Psychomotor development is mostly normal, although deterioration has been observed in the later phases of this disorder. Very severe forms, with corneal clouding, hepatosplenomegaly, marked histiocytosis, with death before 6 months of age (type 4) or death in utero have been reported.
Pathophysiology
Ceramide is formed during the catabolism of all sphingolipids within the lysosomes and the deficiency of acid ceramidase leads to the accumulation of ceramide exclusively in lysosomes in most tissues. Extremely high levels of ceramide have been observed in the urine, but it is not increased in the plasma of patients. The accumulated intralysosomal ceramide does not appear to interfere directly with the regulatory roles of ceramide and its derivatives in other subcellular compartments, e.g. in apoptosis. The accumulation of ceramide may alter membrane fluidity and raft formation and, consequently, receptor-mediated signalling.
Genetics
The acid ceramidase gene (ASAH1) has been cloned and about 20 mutations have been identified in patients. It is not possible to make any deductions about a genotype–phenotype correlation because of the small number of patients analysed. The clinical severity does not correlate with the residual activity measured under non-physiological conditions, but there is a good correlation with the level of lysosomal storage of ceramide.
Diagnosis
Historically Farber disease was diagnosed by the measurement of the accumulation of ceramide in cultured fibroblasts either by including [14 C]stearic acid-labelled sulfatide or sphingomyelin in the medium for 1–3 days or by the enzymic determination of extracted, unlabelled ceramide. Acid ceramidase activity can also be measured in leukocytes and cultured fibroblasts using synthetic substrates in the presence of added detergent. Recently a simple fluorigenic assay using a synthetic substrate incorporating umbelliferone has been developed for the measurement of acid ceramidase activity in cells. It should make the diagnosis of Farber disease more accessible and supersede the radioactive methods. A novel mass spectrometric method for quantifying sphingolipids in extracts of cultured fibroblasts may also be applicable to the diagnosis of Farber disease. Carrier detection is based on mutation analysis. Prenatal diagnosis has been carried out by measuring the ceramidase activity in CV and CAC or by lipid-loading tests in CAC. Genetic testing is the method of choice when the family mutations are known.
Treatment
There is improvement in the peripheral manifestations of infantile Farber disease after bone marrow transplantation but neurological deterioration continues even in mildly symptomatic patients. However, in patients without neurological involvement, allogeneic stem cell transplantation results in almost complete resolution of granulomas and joint contractures and considerable improvement in mobility and joint motility. Gene therapy is in the preclinical stage in large animals using Farber patient haematopoietic cells transduced with a lentivirus vector that overexpresses human acid ceramidase.
Acknowledgement
The authors would like to thank Prof. Thierry Levade for reviewing this section.
Lysosomal acid lipase deficiency: Wolman Disease and Cholesteryl Ester Storage Disease (OMIM: 278000)
A deficiency of the lysosomal enzyme acid lipase (LAL) (EC 3.1.1.13) leads to two main phenotypes, Wolman Disease and Cholesteryl Ester Storage Disease (OMIM: 278000). There is a complete absence of acid lipase activity in Wolman disease, or primary familial xanthomatosis with involvement and calcification of the adrenals. It is a rapidly progressing disease marked by severe failure to thrive, diarrhoea, vomiting and hepatosplenomegaly evident in the first few weeks of life with death usually occurring within 6–8 months from cachexia, complicated by peripheral oedema. Most patients have calcification of the adrenals, which can be detected readily by radiology. Foam cells are found in the bone marrow and later in peripheral blood. The organs contain cells loaded with neutral lipids, especially cholesterol esters and triglycerides, but the levels of cholesterol and triglycerides are normal in plasma.
The presence of some residual acid lipase activity results in the more attenuated disease, cholesteryl ester storage disease (CESD), with a wide spectrum of clinical presentation. It is characterized by liver enlargement, which may be the sole symptom early on and for many years, short stature, chronic gastrointestinal bleeding, chronic anaemia, headaches and abdominal pain. The hepatic fibrosis often leads to atherosclerois. There is not usually any calcification of the adrenals but patients may have sea-blue histiocytosis. Some patients live to adulthood with unpredictable presentation, making diagnosis relatively difficult, whereas others die in their juvenile years. Levels of cholesterol esters are markedly elevated in the liver, whereas the levels of triglycerides are only moderately elevated. There is hyperlipidaemia with a marked decrease in plasma high-densitylipoprotein (HDL) and a slight elevation of liver enzymes.
Pathophysiology
The failure to release cholesterol from cholesteryl esters due to a deficiency of LAL results in upregulation of endogenous cholesterol synthesis and expression of the LDL-receptor gene. There is also increased synthesis of apo-B lipoproteins, which increases delivery of lipoproteins into the lysosomes, particularly in the liver. The clinical features of Wolman disease and CESD result from the deposition of cholesteryl esters and triglycerides in tissues and secondary effects such as the toxicity of lipoprotein oxidation.
Genetics and epidemiology
Lysosomal acid lipase is encoded by the LIPA gene, which is localized on chromosome 10(10q23.2-q23.3) and has been cloned, permitting identification of mutations in Wolman and CESD patients (see Lugowska and Tylki-Szymanska, 2012, for list). In general those mutations that abolish acid lipase activity are found in Wolman disease and those that result in some residual activity are found in CESD. There are no recurrent mutations in Wolman disease, which is very rare in the general population with a frequency as low as 1 in 350,000 births. However, in the Iranian – Jewish community (in which the disease was first described) an incidence as high as 1 in 4,200 newborns has been predicted by screening for carriers of a specific mutation, pG87V. The common splice site mutation del254–277 found in CESD patients produces residual LAL activity, which is sufficient even in heterozygosity to prevent the severe Wolman phenotype. The frequency of CESD among newborns in Germany has been predicted to be 25 cases per million from measurement of the frequency of the common mutation, del254–257 in the population and an estimate of the frequency of this mutation in all cases of CESD. This suggests that CESD is underdiagnosed because this frequency is higher than reported in the literature.
Diagnosis
There is a marked deficiency of acid lipase activity (EC 3.1.1.13) in all tissues of patients with both Wolman disease and CESD. Diagnosis is made by demonstrating a deficiency of acid lipase in leukocytes or cultured skin fibroblasts using a variety of substrates, including radiolabelled triglycerides and cholesterol esters as well as synthetic fatty acid esters of 4-methylumbelliferone and p-nitrophenol. The diagnosis should be confirmed by mutation analysis if possible. Prenatal diagnosis is possible by direct enzyme assay of foetal samples using the synthetic substrates or radiolabelled cholesterol oleate, but molecular testing is the preferred method if the disease-causing mutations have been identified in the family.
Treatment
There are several reports of successful haematopoietic stem cell transplantation in cases of Wolman disease by BMT or unrelated umbilical cord blood transplantation. Based on encouraging pre-clinical data from ERT in acid lipase-deficient rats and mice, trials of enzyme replacement therapy in both Wolman disease and CESD patients are in progress using human recombinant enzyme produced in eggs.
Defects in equilibrative nucleoside transporter 3 – a new lysosomal transporter defect?
Mutations in the human equilibrative nucleoside transporter 3 (ENT3/SLC29A3 gene) have been found in patients with familial Rosai–Dorfman disease, Faisalabad histiocytosis, H syndrome and pigmented hypertrichosis with insulin-dependent diabetes (PHID), all of which are characterized by histiocytosis. Macrophages, which cannot synthesize nucleosides de novo, are highly phagocytic and need to redistribute the nucleosides derived from the lysosomal breakdown of phagocytosed DNA/RNA. It is postulated that a defect in lysosomal ENT3 and the accumulation of nucleosides in the lysosomes could initiate a cascade of secondary events leading to the altered macrophage function observed in these disorders. However, ENT3 has been localized to the mitochondrial as well as the lysosomal membrane and its role in the intracellular transport of nucleosides is not fully understood.
Defects in the lysosomal proteases, cathepsins
At least 15 genetically distinct lysosomal proteases or cathepsins have been described. Most have a catalytic cysteine residue in the active site and are called cysteine proteases, but cathepsins C and E and cathepsins A and G have catalytic aspartic acid and serine residues, respectively. They are predominantly endopeptidases, i.e. cleave the polypeptide chain at internal sites, but cathepsins C (dipeptidyl peptidase I) and X are true exopeptidases and cathepsins B and H have both exo- and endopeptidase activities. Cathepsins are found in all vesicles of the endocytic pathway with some also showing tissue- or cell- or vesicle-specific expression, e.g. cathepsin E in endosomes, cathepsin G in azurophilic granules in neutrophils, whereas cathepsins B, C, H and L are expressed ubiquitously. Cathepsins may be secreted from certain cells for specific functions such as turnover of the extracellular matrix. In addition to the lysosomal catabolism of proteins, specific cathepsins play a role in antigen processing, processing of proteins at various points in the endosomal/lysosomal pathway, and in apoptosis. Other non-cathepsin lysosomal proteases have also been described.
Collectively the lysosomal proteases have the capacity to degrade all polypeptides to their constituent amino acids and possibly to a few dipeptides. A general failure in the lysosomal breakdown of proteins has not been reported, presumably because a deficiency of one cathepsin can be compensated for by another cathepsin due to redundancy in substrate specificity. Very little is known about the specific substrates for cathepsins, and synthetic or purified peptides have usually been used for substrate specificity studies. However, deficiencies of single cathepsins are associated with diseases, reflecting the failure to hydrolyse specific, unknown, polypeptide substrates: cathepsin C in Papillon–Lefèvre syndrome, cathepsin D in NCL10, cathepsin K in immune disorders, and cathepsin K in pycnodysostosis.
Undoubtedly defects of cathepsins and other lysosomal proteases will be shown to contribute to a wide range of diseases because of their involvement in so many key cellular processes unrelated to bulk protein turnover in the lysosome. The potential multifunctional role of cathepsins is illustrated by the deficiency of cathepsin A in galactosialidosis, where cathepsin A has a stabilizing role in the formation of a multiple enzyme complex unrelated to its proteolytic activity (see Chapters 15). As the predominant biochemical feature of galactosialidosis is a defect in the catabolism of glycoproteins, it has been classified as a glycoproteinosis.
Disorders of biogenesis of lysosome-related organelles
(Contributed by Timothy M Cox)
Numerous genetic defects affect protein complexes implicated in the biogenesis and function of lysosomes and their related secretory organelles such as melanosomes; partial albinism is thus a common feature. Several distinct organelles with specialized functions closely resemble lysosomes and are known as lysosome-related organelles. These include δ-granules in platelets; Weibel–Palade bodies of endothelial cells; lytic granules and vesicles implicated in the immune “synapse” in lymphocytes; basophil and azurophil granules in polymorphonuclear leucocytes; lamellar bodies in type 2 pneumocytes; neuromelanin granules in the catecholaminergic neurones of the nigro-strial pathway, and the melanosomes of the iris, choroid and skin. Most of these organelles are also maintained at an acidic pH and have a membrane composition with many similarities to those of lysosomes.
Disturbances of the endosome–lysosome vesicular trafficking network occur at several distinct steps. Disrupted vesicular traffic observed in these albinism syndromes resembles some of the consequences of other lysosomal disorders but there is additionally defective biogenesis of lysosomes and abnormal trafficking of allied organelles and their related proteins – sometimes with impaired extrusion of specialized granules or their contents. These disorders most characteristically affect melanosomes and platelet dense granules but may or may not affect the other related organelles. Pigmentary abnormalities, including the distribution of pigment in hair shafts (as in Chédiak–Higashi syndrome), and impaired platelet function and/or structure are often the first indication of the diagnosis.
Genetic defects in the processes required for the organization of membranes and trafficking of vesicles that lead to the formation of functional lysosomes and lysosome-related organelles, also illustrate their common biogenesis – and reveal much about their complex molecular physiology in differentiated cells.
Hermansky–Pudlak disease(s)
The Hermansky–Pudlak syndrome (HPS) is a group of autosomal recessive disorders affecting specialized secretory organelles. HPS is characterized by a bleeding tendency due to abnormal platelets; reduced pigmentation of the skin, iris and hair – and diverse inflammatory complications including granulomatous colitis, cardiomyopathy and severe fibrosis of the lung. The basic cause of this group of disorders is mis-sorting of molecules typically destined for lysosomes or related organelles; albinism reflects mis-targeting of the tyrosinase-related proteins either to early endosomes or if they are delivered to the melanosome, defective exocytosis leads to their pathological retention. There is accumulation of ceroid lipofuscin pigment in lymphocytes, monocytes, and macrophages in bone marrow. In the lung, defective release of surfactant phospholipid by type 2 pneumocytes leads to inflammatory injury, emphysema and pulmonary fibrosis. Deficient secretion of ADP, which promotes platelet clumping, occurs because the platelet δ-granules are absent: this causes easy bruising, bleeding in the mucous membranes and severe bleeding after injuries and surgery. In women there may be excessive menstruation and bleeding complications in pregnancy and childbirth. Oculocutaneous albinism results from defective formation of melanosomes.
Clinical diagnosis is suggested by the occurrence of colitis or granulomatous colitis often masquerading as Crohn’s disease in patients with fair skin, blue irides and white or blonde hair; increased pigmentation of the hair may occur after puberty. These patients frequently have a tendency to bruise or bleed with prolonged bleeding time and abnormal platelet-function tests. Bone marrow examination shows pigmentary granules in macrophages and autofluoresence due to ceroid material in urinary sediment, and autofluorescent material in oral mucosa biopsy specimens may be detected. Chronic exposure to light is strongly associated with skin injury and cancer; they may also have moderate visual impairment with jerky eye movements due to nystagmus and ocular photophobia. Fortunately, blindness is rare and loss of vision does not usually progress beyond childhood. In later life, the occurrence of fatal lung fibrosis with respiratory failure may alert physicians to the diagnosis of this autosomal recessive disease, sometimes confirmed by a careful family history.
Nine genetically distinct types of Hermansky–Pudlak syndrome have been identified. The affected proteins in type 1 and types 3–9 belong to the biogenesis of lysosome-related organelles complexes 1–3 (BLOC1–3). Definitive diagnosis of these forms of HPS relies upon molecular analysis of genes of the members of the BLOC1–3 families. Hermansky–Pudlak type 2 (HP2), has been shown to be due to mutations in the ADTB3A gene, which encodes the β3A subunit of the adaptor protein complex AP3 involved in the formation of vesicles between the trans-Golgi network and late endosomes. Patients with HPS2 have an immunodeficiency due to congenital neutropenia, as well as platelet defects and partial albinism.
HPS is very rare in most populations, affecting 1 in 500,000 to 1 in 1,000,000 individuals worldwide. Type 1 is more common in northwest Puerto Rico, where the prevalence is about 1 in 1,800 persons; Hermansky–Pudlak syndrome type 3 is also frequent in central Puerto Rico, in Japan, and in an isolated community in Switzerland.
Chédiak–Higashi syndrome
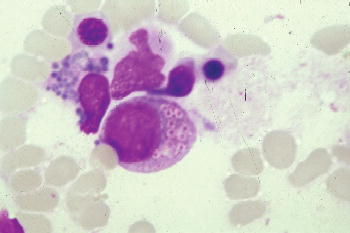
This disorder is also characterized by oculocutaneous albinism, easy bruising and bleeding caused by defective platelet dense bodies but recurrent infections, with neutropenia, impaired chemotaxis and bactericidal activity and abnormal natural killer-cell function, also occur. Most patients experience a phase with lymphohistiocytic infiltration of multiple organs, which resembles lymphoma and appears to be induced by Epstein–Barr viral infection. Death often occurs in childhood from infection, bleeding, or development of this accelerated phase of illness. Chédiak–Higashi syndrome is suggested by partial oculocutaneous albinism, photophobia, nystagmus, large eosinophilic, peroxidase-positive inclusion bodies in the myeloblasts and promyelocytes of the bone marrow, neutropenia, abnormal susceptibility to infection, and development of malignant lymphoma. Death often occurs before the end of the first decade.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


