2 Department of Biochemistry, Children’s Hospital, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
16.2 Multiple Sulfatase Deficiency
16.1 Defects in Transport: Mucolipidosis II alpha/beta, Mucolipidosis III alpha/beta and Mucolipidosis III gamma
Mucolipidosis type II (ML II) and mucolipidosis type III (ML III) are autosomal recessive disorders caused by defects in the N-acetylglucosamine-1-phosphotransferase (GlcNAc-1-phosphotransferase) complex, which is composed of three subunits, α, β and γ. Mutations in the gene encoding the alpha/beta subunits (GNPTAB) lead to ML II alpha/beta, which was also called I-cell disease (OMIM #252500), or to the clinically milder condition, ML III alpha/beta (OMIM #252600), which was formerly called pseudo-Hurler polydystrophy (ML IIIA – Table 16.1). ML III gamma (OMIM #252605), which was formerly called variant ML IIIC, arises from mutations in the gene encoding the γ subunit of GlcNAc-1-phosphotransferase (GNPTG) [1]. The GlcNAc-1-phosphotransferase complex catalyses the first step in the generation of the mannose 6-phosphate (Man6P) recognition marker required for efficient targeting of soluble lysosomal proteins (acid hydrolases and activator proteins) to lysosomes [2].
Case histories
Epidemiology
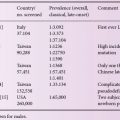
ML II and ML III are rare lysosomal storage disorders with a variable prevalence of 0.16 (The Netherlands), 0.22 (Czech Republic), 0.31 (Australia), 0.4 (Japan) or 0.8 (north Portugal) per 100,000 live births for ML II. The highest ML II frequency, estimated at 16.2/100,000, has been found in Sanguenay-Lac-Saint-Jean (Quebec, Canada). The prevalence of ML III was reported to be 0.08 and 1.89 per 100,000 in The Netherlands and north Portugal, respectively.
Genetic basis
The GlcNAc-1-phosphotransferase complex is composed of three different subunits α, β, and γ in a molar ratio of 2:2:2 and is encoded by two genes. The GNPTAB gene, which is located at chromosome 12q23.3, encodes a 145 kDa α/β subunit precursor [3] that is catalytically activated by proteolytic cleavage by the site-1 protease involved in the cholesterol homeostasis [4] (Figure 16.3 [5]). The GNPTG gene, which is located at chromosome 16p13.3, encodes the soluble 36 kDa γ subunit of unknown function [6]. ML II alpha/beta and ML III alpha/beta patients carry mutations in GNPTAB whereas ML III gamma patients have mutations in the GNPTG gene.
Table 16.1 Classification of mucolipidosis types II and III.
| Former nomenclature | New nomenclature | |
| I-cell disease | ML II | ML II alpha/beta |
| pseudo-Hurler Polydystrophy | ML IIIA | ML III alpha/beta |
| ML III variant | ML IIIC | ML III gamma |
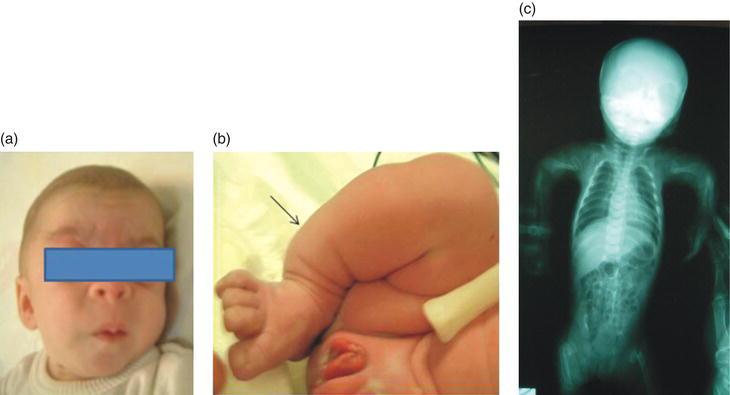
Figure 16.1 Patient with ML II alpha/beta. (a) Coarse facial features, long philtrum and antevarted nares. (b) Bowed leg. (c) X-ray of the spine showing severe hip dysplasia, and dysostosis multiplex of the lower limbs bones.
Pathophysiology
Defects in GlcNAc-1-phosphotransferase lead to missorting of multiple lysosomal hydrolases in mesenchymal cells especially fibroblasts and the lysosomal accumulation of different non-degraded macromolecules, lipids, amino acids or sugars (e.g. glycosaminoglycans, GM2-gangliosides, cholesterol, cystine or sialic acid). Many other cultured cells such as hepatocytes, Kupffer cells, and leukocytes exhibit normal levels of lysosomal enzyme activities. Therefore, peripheral leukocytes are not suitable for ML II and ML III diagnostics. In addition, in liver, kidney, spleen, and brain tissue of ML II patients nearly normal activities of lysosomal enzymes are detectable. The cellular consequences of the accumulating material are still unclear and appear to depend on the composition and extent of storage and the type of storing cells.
Clinical presentation
Neonates with ML II alpha/beta might show craniofacial abnormalities with striking gingival hypertrophy. Importantly, patients with clinical phenotypes intermediate between the ML II alpha/beta and ML III alpha/beta phenotypes have been reported with slower progression than ML II alpha/beta and more severe progression than ML III alpha/beta phenotypes (Table 16.2).
Figure 16.3 Organisation of the GNPTAB and GNPTG genes and their encoded α/β subunit precursor membrane protein (1,256 amino acids) and the soluble γ subunit (305 amino acids), respectively. The α/β subunit precursor is cleaved by the Golgi-resident site-1 protease into mature and enzymatically active α and β subunits (modified from Kollmann et al. 2010 [5]).
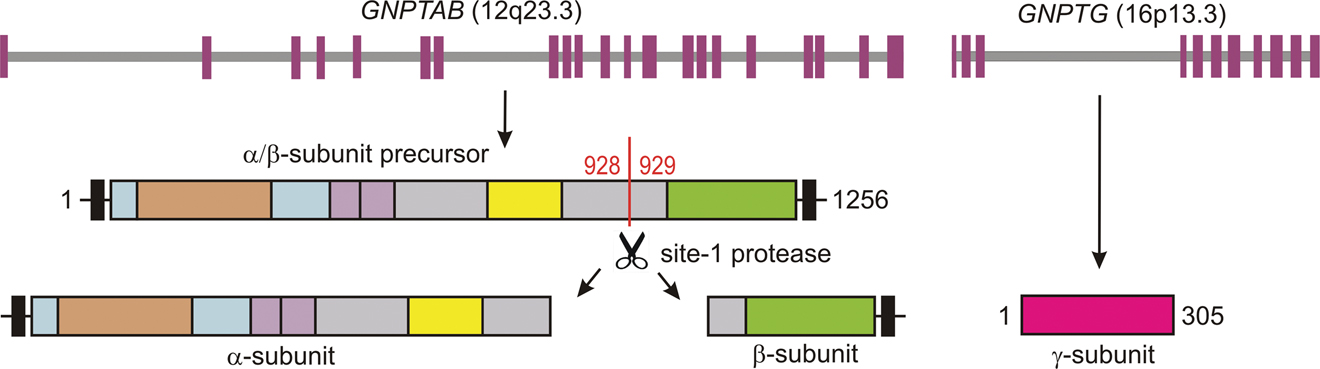
Table 16.2 Clinical fi ndings and symptoms of mucolipidosis types II and III.
| Typical time of onset | Disorder | Signs and symptoms |
| Prenatal | ML II alpha/beta | Short and bowed lower limbs; hips and knees contractures; bone fracture; transient alveolomaxillary defect (TAD); osteopenia; periosteal new bone formation (“cloaking”). |
| Postnatal | ML II alpha/beta | Dysmorphism: coarse facies; gingival hypertrophy, short neck Neurology: Developmental delay; intellectual disabilities; hypotonia Eyes: corneal clouding Skeletal: short bowed limbs; joint contractures; pectus carinatum; thoracal asymmetry; congenital hip dislocation; osteopenia Abdomen: hepatosplenomegaly; umbilical hernia; inguinal hernia Heart: mitral valve thickening; dilated cardiomyopathy transient neonatal hyperparathyroidism |
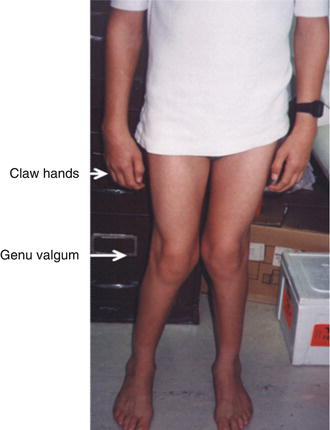
| Early infancy and adulthood | ML III alpha/beta & ML III gamma* | Skeletal: progressive joint stifness; stiffness of the small joints; claw hand; carpal tunnel syndrome; tarsal tunnel syndrome; scoliosis; kyphosis; hyperlordosis; genu valgum; shoulder, decreased mobility and/or contractures of knees and hip joints; upper limb paraesthesia; progressive growth retardation; C1-C2 instability; progressive bone dystrophy X-rays: progressive development of “J” shaped sella turcica; oar-shaped ribs; anterior inferior beaking of lower thoracic to upper lumbar vertebral bodies; flared iliac wings; progressive dysplasia of the femoral heads and necks; coxa valga; “bullet-shaped” proximal phalanges and central pointing of proximal metacarpals Eyes: hyperopic astigmatism and corneal clouding; retinal and optic nerve abnormalities Heart: aortic and /or mitral valve thickening; dilated cardiomyopathy Abdomen: umbilical hernia; inguinal hernia |
*The ML III gamma patients seems to have a milder clinical picture than the ML III alpha/beta patients.
ML III alpha/beta and ML III gamma are slowly progressive disorders mainly affecting skeletal, joint and connective tissues. Clinical onset is in early childhood with restricted joint mobility, and skeletal changes that mainly affect the pelvis and spine. ML III gamma seems to be less severe than ML III alpha/beta.
Natural history
ML II alpha/beta is a severe disease with onset of signs and symptoms at birth and a fatal outcome most often in early childhood. ML III alpha/beta patients show first clinical symptoms at approximately 3 years of age whereas data on life expectancy are missing. The onset of first symptoms in ML III gamma patients is in infancy as well and they are known to survive to the fourth or fifth decades; the oldest reported patient was 86 years old [7]. ML III might be misdiagnosed as rheumatoid arthritis.
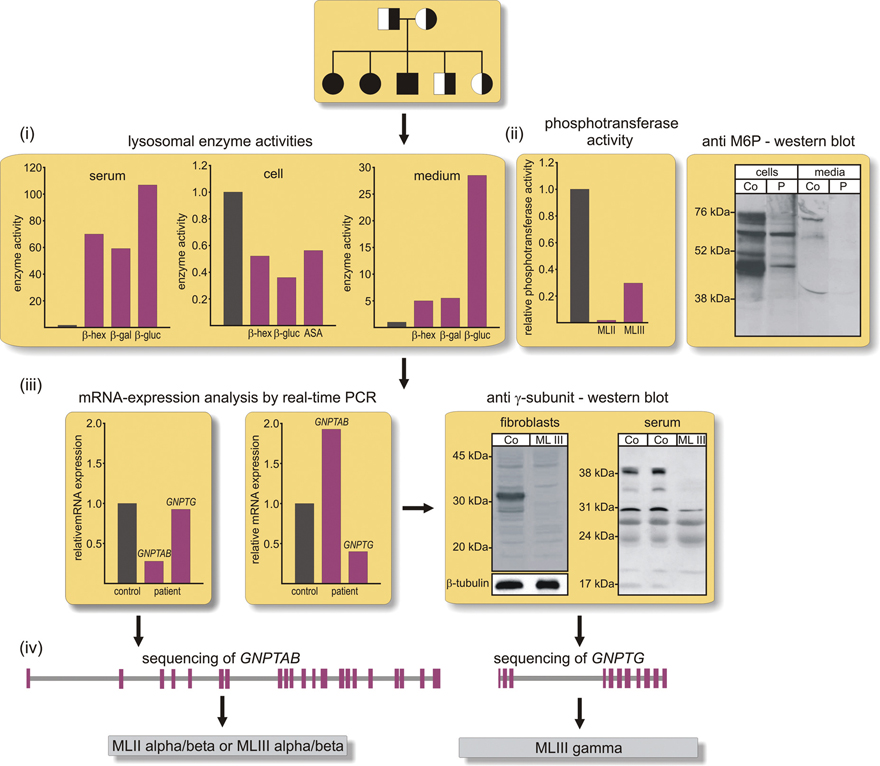
Figure 16.4 Diagnostic pathway for mucolipidosis types II and III. The activities of a set of lysosomal enzymes, e.g. β-hexosaminidase (β-hex), β-glucuronidase (β-gluc), β-galactosidase (β-gal), and arylsulfatase A (ASA) should be measured in the serum, in extracts of cultured fibroblasts and conditioned media. In comparison to controls (black column) the activities of the lysosomal enzymes are decreased in patient cells but increased in serum and conditioned cell media, demonstrating the missorting of lysosomal enzymes. (ii) Defects in the generation of the Man6P recognition marker can be analyzed by radioactive measurement of GlcNAc-1-phosphotransferase activity in extracts of cultured cells, or by Man6P-western blotting of cell extracts and aliquots of conditioned media. Both experimental approaches support the diagnosis of ML II or ML III. (iii) Since many of the known mutation in GNPTAB or GNPTG lead to frameshift and unstable transcripts due to “nonsense-mediated mRNA decay”, the mRNA expression of GNPTAB/GNPTG is performed in fibroblasts. Reduced GNPTG mRNA levels have been reported to be accompanied by compensatory up-regulation of GNPTAB mRNA. The decreased expression of GNPTG mRNA can be confirmed by western blotting for the human γ subunit. (iv) The direct sequencing and DNA analysis of the GNPTAB or GNPTG genes allow the diagnosis of ML II alpha/beta or ML III alpha/beta or gamma.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


