Epidemiology and clinical presentation
Given the rarity of this disease, there is no comprehensive survey of the demographics and disease spectrum for galactosialidosis. All patients have clinical manifestations typical of a lysosomal disorder such as coarse facies, cherry-red spots, vertebral changes, foam cells in the bone marrow and vacuolated lymphocytes. Three phenotypic subtypes are recognized. The early infantile form is associated with foetal hydrops, edema, ascites, visceromegaly, skeletal dysplasia and early death. Hepatosplenomegaly, growth retardation, cardiac involvement and rare occurrence of important neurological signs characterize the late-infantile type. The majority of reported patients belong to the juvenile/adult group and are mainly of Japanese origin. Myoclonus, ataxia, angiokeratoma, mental retardation, neurological deterioration, absence of visceromegaly and long survival are quite characteristic of this subtype.
Table 15.1 Galactosialidosis patients reported in the literature.
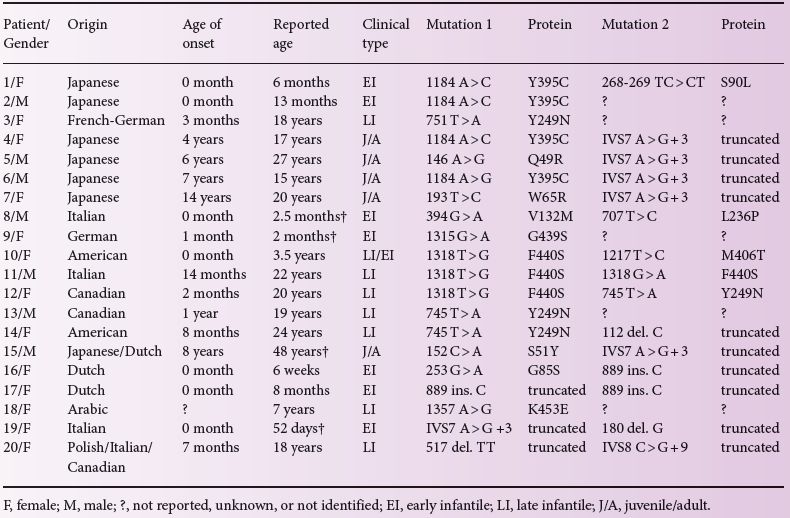
Figure 15.1 Genomic organization of the CTSA gene. Boxes represent exons 1 through 15; exons 1 and 15 contain UTRs. Two intronic splice mutations and 13 amino acid substitutions caused by missense exonic mutations are indicated below and above the gene, respectively.
Genetic basis
The gene encoding PPCA (CTSA, Genomic coordinates (GRCh37): 20:44 519 590–44 527 458) is located on chromosome 20q13. The CTSA gene overlaps at its 5′and 3′ ends with two other genes both transcribed from the antisense strand relative to CTSA. The gene at the 3′ end (PLTP) encodes a phospholipid transfer protein whereas that at the 5′ end (NEURL2) encodes OZZ, a striated muscle-specific E3-ubiquitin ligase. Nineteen disease-causing mutations have been identified in the CTSA gene, the majority of which are missense mutations resulting in single amino acid substitutions (Table 15.1 and Figure 15.1). Other reported mutations include nonsense mutations, frameshift mutations or absence of the CTSA mRNA leading to either premature termination of the translated protein and a truncated protein product or the complete lack of the PPCA protein. The patients are either homozygous or compound heterozygous for the CTSA mutations (Table 15.1). Their parents are non-symptomatic heterozygous carriers. Generally, there is a good correlation between the type of CTSA mutation, the level of residual cathepsin A enzyme activity and the severity of the clinical phenotype. Mutations that result in up to 7% residual enzyme activity are typically associated with the juvenile or juvenile/adult phenotype (Table 15.1). Several of the identified amino acid substitutions were modelled into the tertiary structure of the PPCA precursor: none affected the catalytic machinery, but rather disrupted the integrity and stability of the enzyme. Notably two engineered active site mutations were shown not to affect the protective properties of the enzyme towards the two glycosidases (see below). This finding suggested that if such genetic mutations were to occur naturally, they would not result in a galactosialidosis phenotype.
Biochemistry
PPCA is a serine carboxypeptidase, which is synthesized as a 54-kDa zymogen and processed in lysosomes into a two-chain mature form of 32 and 20 kDa after endoproteolytic removal of a 2-kDa internal peptide. The enzyme carries two N-linked glycans, one on each chain, and is routed to the lysosome via the mannose-6-phosphate receptor pathway. PPCA has the unique capacity to specifically bind to β-GAL and NEU1 shortly after synthesis. Association of the three proteins in an early biosynthetic compartment assures the regulated trafficking of the two glycosidases to the lysosome as well as their intralysosomal activation and stable conformation in a multi-enzyme complex. Besides its chaperone-like function and protective properties, PPCA is catalytically active at both acidic and neutral pH and functions as cathepsin A/deaminase/esterase on a subset of neuropeptides, including substance P, oxytocin and endothelin-I. In a recent study multiple binding sites on the surfaces of both PPCA and NEU1 were identified that appear to be essential for the interaction of the two enzymes. One of these PPCA-binding sites on NEU1 can also bind to other NEU1 molecules, albeit with lower affinity. Therefore, in absence of PPCA, NEU1 tends to self-associate into chain-like oligomers and form insoluble aggregates. Thus, PPCA prevents aberrant self-association of NEU1 by promoting the formation of a PPCA–NEU1 heterodimeric complex. The proposed mechanism of interaction between NEU1 and PPCA provides a rationale for the secondary deficiency of NEU1 in galactosialidosis. In addition to a complete loss of NEU1 and a partial loss of β-GAL and N-acetylgalactosamine-6-sulfate-sulfatase activities, cathepsin A activity was found to be completely or partially deficient in cultured fibroblasts or lymphocytes of all tested patients with galactosialidosis.
Pathophysiology
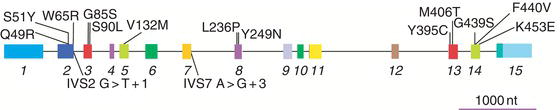
Galactosialidosis belongs to the glycoproteinosis subgroup of lysosomal storage diseases (see Chapter 14) and affects primarily the reticuloendothelial system. In patients with this disease the secondary NEU1-deficiency, which preferentially targets the cleavage of α2-3- and α2-6-linked sialic acid residues on glycoproteins, leads to the accumulation of N-acetyllactosamine-type sialyloligosaccharides and glycopeptides in multiple tissues and body fluids. Fibroblasts and urine of galactosialidosis patients contain fully sialylated di- and triantennary N-linked oligosaccharides, and completely lack terminal galactose residues. Ultrastructural and histochemical analysis of patient-derived hepatocytes and Kupffer cells in the liver, glomerular and tubular epithelial cells in the kidney, as well as cultured fibroblasts has revealed the presence of numerous membrane-bound vacuoles filled with storage material. The severe secondary deficiency of NEU1, rather than the partial deficiency of β-GAL, is thought to be the primary contributing factor to the pathophysiology of galactosialidosis. This is supported by the fact that patients with galactosialidosis share several of the key clinical and biochemical features of patients with the lysosomal storage disease sialidosis (OMIM 256550), caused by structural lesions in the NEU1 gene. In contrast, patients with GM1-gangliosidosis and Morquio B syndrome (OMIM 230500, 230650, 230600, 253010), with a primary defect in β-GAL, develop a glycosphingolipid storage disease that affects primarily the central nervous system, and a systemic disease without central nervous system involvement, respectively.
Diagnosis
The diagnosis of galactosialidosis can be suspected in patients with clinical features of a lysosomal storage disease who show Hurler-like facial features, growth retardation, sialyloligosacchariduria and vacuolated lymphocytes in their peripheral blood. The demonstration of a combined deficiency of β-GAL and NEU1 in lymphocytes and/or cultured skin fibroblasts is the preferred method of biochemical diagnosis. Mutation analysis based on DNA sequencing of the exons and the intron–exon junctions unequivocally establishes the diagnosis when a mutation is detected. In non-immune hydrops foetalis pregnancies, or when both parents are known heterozygous carriers, prenatal diagnosis may be performed by measuring cathepsin A and neuraminidase activities, or by DNA mutation analysis on either chorionic villi, amniotic fluid, and cultured amniotic cells.
Figure 15.2 H&E staining of tissue sections from galactosialidosis (Ctsa-knockout) mice showing extensive vacuolization of different cell types in multiple organs compared to the corresponding wild-type sections. Magnification 40 ×.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


