Epidemiology
Data about the epidemiology of MPSs is available only for few countries and regions. Although MPSs, as a group, are estimated to occur in one in every 22,000 individuals, individual prevalence of each MPS is much lower. The relative frequency of different MPS types also differs from region to region, but MPS II seems to be the most common in most places, followed by MPS III (A and B), MPS I and MPS IV A. MPS VI is rare in Europe and North America, but relatively frequent in some countries as Brazil and Turkey. MPS VII, MPS IV B and MPS III (C and D) are much rarer, and MPS IX has been described in only two families so far. Although it is believed that the majority of the severe cases of MPS are identified, usually after a long diagnostic “odyssey”, it is probable that the diagnosis of MPS in some attenuated patients is missed. It is noteworthy that many children with MPS are submitted to surgery before a diagnosis is confirmed.
Genetic basis
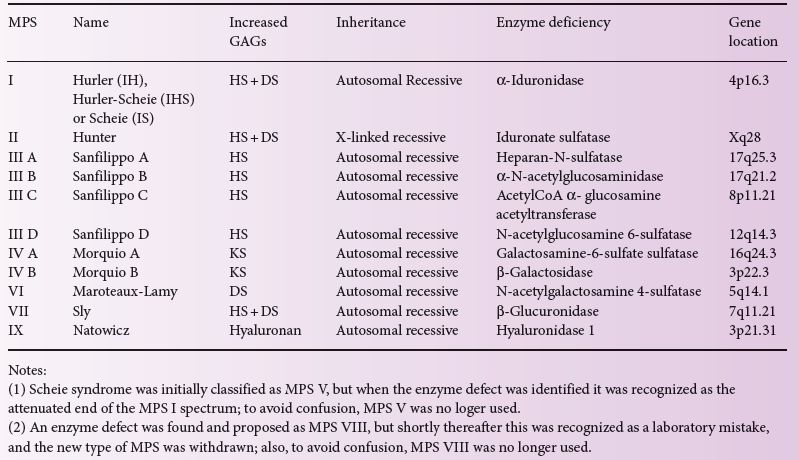
With the exception of MPS II, which is X-linked, all of these diseases are inherited as autosomal recessive traits (Table 12.1). These diseases are genetically very heterogeneous, with many mutations described for each MPS type. In general, there is a trend that nonsense and frameshift mutations lead to more severe disease, while missense mutations are associated with more attenuated pictures, but it is usually difficult to predict phenotype from the genotype on an individual basis. All the MPS types, including MPS II, are true recessive diseases, with no detectable manifestations in carriers. A few females with MPS II were reported, these being extremely rare occurrences not related to carrier status but to major rearrangements and/or skewed inactivation.
Table 12.1 The classification of the mucopolysaccharidoses.
Pathophysiology
Although MPS storage plays an important role in the pathophysiology of MPSs, it is now known that it is not the only pathogenic mechanism involved in these diseases. While some clinical signs, such as hepatosplenomegaly, are caused directly by the storage of partially degraded GAGs within the lysosomes, these molecules are also located outside of the acidic organelle. Therefore they can activate inflammatory pathways and an innate immune response via the toll-like receptor 4 and the complement system, which might contribute to some aspects of these diseases, including neuroinflammation, short bones and aortic fragmentation. The undegraded GAGs can also upregulate the expression of destructive proteases and induce apoptosis in tissues such as the joints and the bones.
Clinical presentation
The MPSs are a group of heterogeneous and multisystem disorders, with broad heterogeneity across types. Most cases have disease manifestations which bring major impairment to lifespan and quality of life. Within the same type there are severe and attenuated patients with intermediate cases confirming a continuum of disease severity between the two extremes. From the clinical point of view, MPS cases can be classified into three main groups: patients with predominant “visceral phenotype”, those with predominant “neurodegenerative phenotype” and those with predominant “skeletal phenotype”. The group with predominant “visceral phenotype” includes patients with MPS I, MPS II, MPS VI and MPS VII, who present with the main manifestations of coarse facies, visceromegaly (hepatosplenomegaly), hernia, joint stiffness, upper airway obstruction and heart disease (Figure 12.1). Bone dysplasia, known as “dysostosis multiplex”, and other skeletal deformities are present (Figure 12.2). Although growth may be normal and even increased in the first years, short stature is eventually established, and mental deterioration starting in childhood occurs in the severe cases of MPS I and MPS II and also in MPS VII. Corneal clouding is frequent (except in MPS II), and also hearing loss (marked in MPS II). The group with the “neurodegenerative phenotype” is formed by patients with MPS III. In these patients, the visceromegaly, coarse facies, joint disease and bone dysplasia (dysostosis multiplex) are relatively mild, and the clinical picture is marked by neurodegeneration, which usually starts between 3 and 5 years of age and progresses steadily. Behavioural disturbances and hyperactivity are often reported in the early stages of the disease (Figure 12.3). The group with the “skeletal phenotype” is formed by patients with MPS IV, who show a skeletal dysplasia (different from dysostosis multiplex) with genu valgum, pectus carinatum, kyphoscoliosis, hypoplasia of odontoid with atlanto-axial instability, among other bone problems. Patients are mentally normal and have very short stature, presenting corneal opacity and valve disease. They do not have visceromegaly or coarse facies, and usually show joint laxity instead of joint stiffness (Figure 12.4). MPS IX was not included in any of these 3 phenotypes since in the cases reported the main clinical finding is the presence of joint swellings and synovial masses.
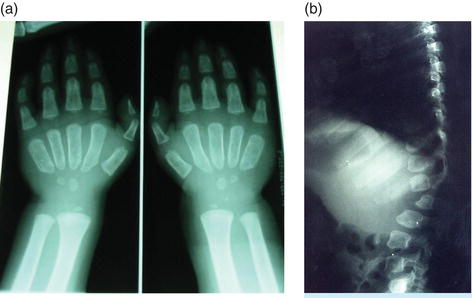
Figure 12.1 Dysostosis multiplex observed in X-rays of (a) hands and (b) spine of a MPS I patient. The broad and short metacarpals are typical, as are the shape of the vertebral bodies.
Related posts:
 10: Types A and B Niemann–Pick Disease
10: Types A and B Niemann–Pick Disease
 18: Neuronal Ceroid Lipofuscinoses
18: Neuronal Ceroid Lipofuscinoses
 11: Niemann–Pick Disease Type C
11: Niemann–Pick Disease Type C
 23: Newborn, High Risk and Carrier Screening for Lysosomal Storage Disorders
23: Newborn, High Risk and Carrier Screening for Lysosomal Storage Disorders
 5: Classification of Lysosomal Storage Diseases
5: Classification of Lysosomal Storage Diseases
 21: Central Nervous System Aspects, Neurodegeneration and the Blood–Brain Barrier
21: Central Nervous System Aspects, Neurodegeneration and the Blood–Brain Barrier
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


