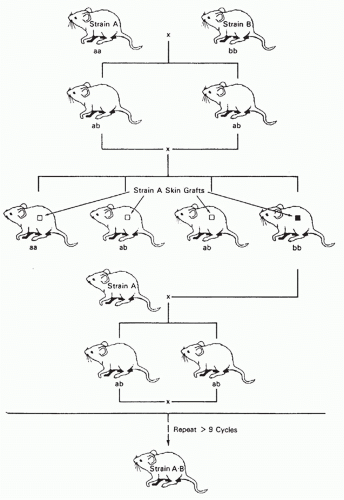
“Acute cellular rejection,” which is characterized by a mononuclear cell infiltrate in the graft, is the most common type of organ allograft rejection. Acute rejection is most common during the first 3 months after transplant, but may occur at any time, especially if immunosuppressive medication is withdrawn. Acute rejection is T cell-dependent, and its treatment, which is usually successful, includes increased doses of standard immunosuppressive drugs or antilymphocyte antibodies.
The use of newer immunosuppressive drugs and anti-T-cell antibodies has markedly reduced acute rejection rates. For example, the vast majority of kidney transplant recipients never experience an episode of acute rejection. It is now quite rare to lose a transplanted organ to cellmediated rejection during the first year after transplantation. However, the use of these highly effective immunosuppressive treatments is associated with significant morbidity.
Experimental models for acute rejection include nonprimarily vascularized skin grafts, heart graft fragments, artificial “sponge” allografts, or islet transplants in rodents, which may not accurately reflect the processes of rejection for primarily vascularized organs. While there are models of heart, kidney, liver, and other types of primarily vascularized organ transplants in rodents, these types of transplants are more tolerogenic and hence more easily accepted than similar transplants in large animals and humans. Studies of primarily vascularized organ transplants in large animals, such as monkeys or pigs, have obvious clinical relevance, but are expensive and require many special resources.
Acute GVHD is the counterpart of cellular rejection that involves graft-versus-host alloreactivity, usually in the context of HCT, but also sometimes with organ transplants that carry significant amounts of donor lymphoid tissue (eg, liver). Like acute rejection, acute GVHD is T cell-dependent. T-cell depletion of the donor hematopoietic cell graft prevents GVHD but is associated with increased rates of graft rejection and relapse of malignant diseases.
Sensitization and Cell Trafficking during Rejection and Graft versus Host Disease
Transplanted tissue contains passenger leukocytes of donor origin that have the characteristics of immature DCs.
169 In response to the inflammatory signals that are triggered by retrieval and transplantation, both within the tissue itself as well as in the recipient,
170 the donor-derived passenger leukocytes rapidly leave the graft and migrate to the secondary lymphoid tissues of the recipient.
171 Secondary lymphoid tissues comprise the spleen, lymph nodes, and gut- or mucosal-associated lymphoid tissue, and depending on the location of the graft, the passenger leukocytes will migrate to the tissue that drains the graft site where they encounter naïve T cells. After transplantation, both in situ within the tissue and during migration, the passenger leukocytes
acquire the phenotypic and functional characteristics of mature DCs, expressing high levels of MHC class I and II molecules as well as other cell surface costimulatory molecules necessary to fully activate naïve CD4+ and CD8+ T cells.
172 Once in the secondary lymphoid tissues they act as professional APCs, presenting antigens expressed in the transplanted tissue to recipient T cells via the direct pathway of allorecognition.
173Naïve T cells recirculate around the body and are constantly moving through the secondary lymphoid tissues sampling the APC, both host- and (after transplantation) donor-derived, for antigen.
174 If a naïve T cell with a TCR that can recognize a donor MHC molecule encounters the donor-derived passenger leukocyte in the draining lymphoid tissue as it recirculates, it will stop, interact, and differentiate into an antigen-experienced effector T cell. In support of the secondary lymphoid tissue being the primary site for sensitization of naive T cells and initiation of rejection after solid organ transplantation, Lakkis and colleagues
175 showed that cardiac allografts were not rejected in splenectomised aly/aly mice that lack secondary lymphoid tissue as a result of a mutation in the gene encoding NF-kB-inducing kinase
176 and suggested that in this situation permanent graft acceptance was due to immunologic ignorance. Other studies supporting the concept that secondary lymphoid tissues draining the graft are the key site for initiation of the immune response have followed the fate of T cells of a known specificity for donor antigen as they respond.
177Similarly, after bone marrow transplantation (BMT) the initiation of GVHD also takes place in the secondary lymphoid tissue, with evidence for the initial proliferation of donor CD4+ T cells followed by CD8+ T cells in secondary lymphoid organs with subsequent homing to the intestines, liver, and skin.
178,179,180 Visualization of T cells responding during the initiation of GVHD showed that while Peyer patches are involved, other secondary lymphoid tissues contribute to the activation of T cells that can home to the gut; mesenteric lymph nodes and spleen are also sites where gut homing T cells were activated.
179In solid organ transplantation, exclusive initiation of rejection in the secondary lymphoid tissues conflicts with the earlier hypothesis that rejection was initiated within the graft itself by donor endothelial cells lining the vessels that could activate T cells directly as they passed through the graft.
181,182 Since these early papers, there have been a number of studies that support this hypothesis. For example, human endothelial cells have been shown to activate naïve T cells in vitro.
183 In the mouse, APCs that are not of hematopoietic origin have been shown to activate CD8+ T cells in vitro and in vivo,
184 thus supporting the concept that T cells may be activated in the graft rather than in the secondary lymphoid tissue. Moreover, splenectomized lymphotoxin α and lymphotoxin β knockout mice that also lack secondary lymphoid tissues were found to reject cardiac allografts, albeit at a slower than normal tempo.
185 Each of these models is subtly different immunologically, and therefore different components of the immune response to an allograft may be differentially affected by the presence or absence of secondary lymphoid tissues. Clearly, in the absence of secondary lymphoid tissue, the initiation of the rejection response by naïve T cells is less aggressive.
While antigen presentation via the direct pathway plays a dominant role in initiating the response to a transplant, a finite number of donor-derived passenger leukocytes is transferred within a transplanted organ. Thus the role of the direct pathway initiated by passenger leukocytes may diminish with time as eventually only “nonprofessional” APCs, including endothelial cells, remain to stimulate direct pathway T cells. Thus the role of endothelial cells within the graft may assume a greater significance with time after transplantation both for the initiation of the response and as a target for direct pathway effector cells. While activation of naïve T cells may occur predominantly in the secondary lymphoid tissues after transplantation, activation of memory T cells in presensitized recipients is quite different. Unlike naïve T cells, memory T cells can migrate to nonlymphoid tissues in the periphery
186 and can trigger rejection through pathways that are independent of secondary lymphoid tissues.
187 Thus, in humans, where there are likely to be both naïve and memory T cells that can recognize or cross-react with donor MHC molecules, rejection may be initiated both within the secondary lymphoid tissue as well as within the allograft by naïve and memory T cells, respectively.
At the same time that donor-derived passenger leukocytes are leaving the graft, recipient leukocytes including APCs are attracted to the graft by the inflammatory mediators and chemokines released in the vicinity of the transplanted tissue. As these cells traffic through the graft, they phagocytose debris arising from tissue damage at the time of transplantation before migrating to the draining lymphoid tissue. The ingested antigens are processed and presented on recipient MHC molecules to T cells in the recipient lymphoid tissue.
188 In addition, soluble antigens released from the graft will also be transported in the blood to the draining lymphoid tissue, where they will be taken up and presented by resident APCs. Common antigenic peptides presented by the indirect pathway are the hypervariable peptide binding regions of MHC molecules.
189 Indirect pathway responses undoubtedly contribute to acute rejection, although the tempo of rejection may be slower due to the lower frequency of T cells that can respond. However, unlike direct pathway allorecognition, the indirect pathway is available for antigen presentation for as long as the graft remains in situ, and therefore becomes the dominant mode of allorecognition long term.
A third pathway of allorecognition has been described more recently, the so-called semidirect pathway that involves the capture of donor MHC-peptide complexes by host APCs. The exchange of fragments of cell membrane between cells that interact with one another is a well described phenomenon in cell biology. In the context of the immune response to an allograft, the transfer of membrane fragments from allogeneic cells expressing donor MHC molecules can result in the presentation of intact donor MHC molecules by recipient APCs to T cells. The significance of the semidirect pathway is still under investigation.
190Traffic of naïve lymphocytes is usually restricted to recirculation between the blood and lymphatic systems. However, once they have been primed in the secondary lymphoid tissues, activated lymphocytes as well as other activated leukocytes must be able to migrate into the graft in order to destroy the transplanted tissue, a process known as leukocyte recruitment.
The inflammatory processes at the site of transplantation generate chemotactic cytokines called chemokines, and upregulation of chemokine receptor expression by activated leukocytes enables them to migrate along the chemoattractant gradient to reach the graft.
191Inflammatory signals also affect blood vessels in the vicinity of the transplant, causing vasodilation and endothelial activation. Activated endothelial cells rapidly externalize preformed granules called Weibel-Palade bodies that contain the adhesion molecule P-selectin
192 and rapidly upregulate expression of vascular cell adhesion molecule and CD62E (E-selectin). At the same time, chemokines released from the graft become tethered to the endothelium, and these alterations in endothelial surface markers advertise to passing leukocytes that an inflammatory process is occurring in the neighboring tissue.
Leukocytes are usually conveyed within the fast laminar flow at the center of blood vessels, but once activated leukocytes reach postcapillary venules in proximity to the graft, they are able to leave this rapid flow and move toward the edge of the vessel. This occurs in response to the local chemokine gradient and is assisted by the slower blood flow in the vasodilated blood vessels near the graft. Leukocyte extravasation is a multistep process. Initially, low-affinity interactions develop between endothelial P-selectin and sialyl-LewisX moieties that are present on the surface of activated leukocytes. These interactions continually form and break down, and the leukocyte “rolls” along the endothelial surface. If chemokines are present on the endothelial surface, conformational changes in leukocyte integrin molecules occur that allow them to bind other endothelial adhesion molecules such as ICAM-1. These higher-affinity interactions cause arrest of the leukocyte on the endothelial surface, allowing it to commence extravasation. Having entered the tissues, the activated leukocytes continue to migrate along chemokine gradients in order to invade the graft.
Antigen Recognition and T-Cell Help in Graft Rejection and Graft-versus-Host Disease
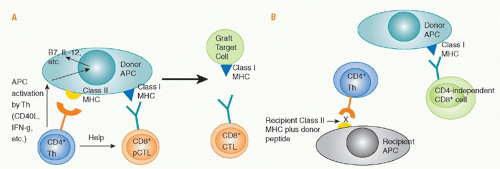
Role of Direct Cluster of Differentiation 4 Allorecognition. Priming of naïve, directly alloreactive T cells requires professional APCs that leave the graft and enter the recipient’s lymphoid tissues. Direct CD4 T-cell sensitization by donor class II MHC antigens may both generate CD4+ effector cells and provide help for the activation, differentiation, and proliferation of cytotoxic CD8+ cells that directly recognize donor class I MHC antigens and destroy the graft (see
Fig. 46.9). Depletion of donor APCs can markedly prolong graft survival,
193,194,195,196,197,198 illustrating the importance of direct allorecognition in inducing rejection. The CD4 help for CD8 cells consists of both cytokine (eg, IL-2) production and “conditioning” of the APC, for example by interactions of CD40 on the APC with CD40L on the activated CD4 cell. These interactions upregulate APC expression of CD80 and CD86 costimulatory molecules and cytokines such as IL-12 and MHC, making the cell a more effective APC. Studies of antiviral immunity indicate that CD4 help is needed for development of full effector function,
199 and for CD8 memory cell survival
200 and function.
201
Studies involving very limited (not clinically relevant) antigenic disparities between donors and recipients suggested that a “three-cell cluster” model involving interactions between helper T cells, effector T cells, and APCs was essential for rejection.
202,203,204,205 However, studies involving more extensive, clinically relevant histoincompatibilities
206,207 suggest that CD4 helper cells sensitized by antigen presented on recipient APCs can provide help for directly alloreactive CD8+ effector cells. It remains possible that a “three-cell cluster” is still essential for CD4 cells to provide help to CD8 cells mediating rejection, and that donor class I MHC/peptide complexes are transferred and picked up by recipient APCs. Recipient APCs with directly alloreactive CD8 T cells would thereby encounter their ligands on the same recipient APC that an indirectly alloreactive CD4 cell recognizes. Such transfer of class I/peptide antigens, resulting in this type of
“semidirect” antigen presentation, requires consideration in transplant models.
208,209,210,211CD4+ T cells alone can cause graft rejection (without CD8 cells) in the setting of class II or multiple minor histoincompatibilities,
198,212,213,214,215 indicating that CD4 T cells can mediate rejection effector functions. In BMT recipients, they can induce GVHD in the absence of CD8 cells in the setting of class II, full MHC, or multiple minor histoincompatibilities,
216,217,218,219 and can reject class II and minor antigen-mismatched bone marrow.
220,221Role of Indirect Cluster of Differentiation 4 Cell-Mediated Allorecognition. Indirectly alloreactive CD4 T cells have roles in skin and solid organ graft rejection,
210,222,223,224,225,226,227,228,229,230,231,232 including the provision of help for class-switched alloantibody responses.
233 This help requires cognate interactions between recipient class II-restricted indirectly alloreactive CD4 cells and host B cells that recognize donor MHC molecules through their immunoglobulin receptors, process them, and present donor MHC peptides with their class II molecules. CD4 cells also contribute to rejection of bone marrow grafts differing only at class I MHC loci, possibly implicating indirect allorecognition.
220,221
Rejection by CD4 cells of skin grafts lacking class II MHC shows the strength of the indirect pathway of rejection.
206,222 Rejection of islet xenografts in mice may depend on indirectly xenoreactive CD4+ T cells.
234 Sensitization of indirect CD4 responses to donor MHC-derived peptides has been demonstrated in patients undergoing graft rejection, and these may be correlated with poor outcomes.
235,236,237,238,239,240,241,242 A major role for indirect allorecognition has been suggested in the setting of chronic rejection,
230,243,244,245,246 in part because of its role in inducing antibody responses, which are implicated in chronic rejection.
247,248,249,250,251 Moreover, the eventual replacement of donor APCs by recipient APCs implicates the latter in long-term graft recipients.
196,241,252,253 Consistently, direct alloresponses tend to subside over time in transplant patients.
254,255,256Nevertheless, donor APC depletion or the lack of donor class II MHC can prevent rejection in some situations.
193,194,195,196,197,198 An essential role for indirect allorecognition has not been demonstrated for acute rejection.
21,253,257,258 Indirectly alloreactive CD4+ cells alone fail to reject skin grafts with minimal class I or minor histoincompatibilities,
202,259,260 or to induce GVHD against class I MHC or minor histocompatibility barriers alone.
216,217 With rodent primarily vascularized allografts, donor APC depletion may, by preventing the strong direct alloresponse, allow the inherent tolerogenicity of the organ to prevail.
Role of Helper-Independent Cluster of Differentiation 8+ T Cells. CD8 T cells can readily reject skin and bone marrow allografts in the absence of CD4 cells,
166,202,220,221,261,262 and alloreactive CD8 T-cell memory can be generated and maintained without CD4 cells.
263 CD8 cells can also induce GVHD without CD4 T cells in the setting of full MHC, class I only, and minor antigen histoincompatibilities.
264,265 Direct recognition of recipient MiHAs on recipient APCs is essential for the induction of CD8-dependent, CD4-independent GVHD in MHC-identical, lethally irradiated mice,
266 but indirect
267 or “semidirect”
211 CD8 recognition of recipient antigens presented by donor APCs amplifies the process.
Together, these studies show that CD4 help is not critical for CD8 cell-mediated rejection or GVHD. However, the requirement for CD4 help may increase in the absence of inflammatory stimuli, as indicated by marked differences in the need for CD4 help for CD8-cell activation and GVL effects in the presence and absence of inflammation.
268,269 Grafts expressing only class I antigen disparities are usually rejected quite slowly, and CD4-independent rejection is relatively easily suppressed by cyclosporine.
270,271,272 Many primarily vascularized grafts that express only a class I antigen disparity require CD4+ cells to initiate rejection, and, when it occurs, CD4-independent rejection by CD8+ cells is dependent on the number of donor APCs in a graft.
166,222 CD4-indepenent CD8+ cells do not reject grafts expressing only a small number of minor antigen disparities and generate only weak helper responses even in the presence of multiple MiHA disparities. CD8+ helper cells also differ from CD4+ helper cells in being unable to provide help for other cell populations.
273 CD8+ cells alone cannot reject skin grafts with only limited class II antigen disparities.
202,218,259,274Cross-Primed Cluster of Differentiation 8 Cells. Peptides of exogenous antigens were originally thought to be presented by MHC class II antigens, whereas those of endogenous cellular antigens are presented by MHC class I molecules.
275,276 However, it is now clear that class I presentation of exogenous peptides (cross-presentation) is essential for many immune responses, including those against microbial and tumor antigens.
277,278,279,280,281 Several pathways have now been delineated for cross-presentation by class I molecules.
282,283,284 CD8 cell crosspriming was originally demonstrated in a transplantation model by Bevan
70 when minor antigen-disparate grafts with MHC antigens of type A were placed on MHC (A × B) F1 recipients and CD8+ cells became sensitized to the minor antigens presented by both A and B types of class I MHC molecules. Activation of cross-primed CD8 cells is strongly dependent on CD4 help and IL-2.
285 Cross-primed CD8 cells recognizing donor MiHAs and MHC-derived peptides are most likely to participate in rejection when there is sharing of class I alleles between the donor and recipient. Without such sharing, the self-class I/allogeneic peptide epitope cannot be presented by the parenchymal or endothelial cells of the graft.
286 However, even without class I sharing, indirect CD8+-cell sensitization can lead to skin allograft rejection, perhaps due to recognition of donor peptides presented by recipient endothelial cells on host-derived vessels that revascularize the graft.
287,288 Cross-primed CD8 cells might also contribute to graft rejection via indirect effector mechanisms upon antigen recognition on host APCs in the graft or by producing inflammatory cytokines.
289 Some of the rejection processes previously attributed to cross-primed CD8 cells may in fact be mediated by CD8 cells seeing intact donor MHC-peptide complexes on recipient APCs (“semidirect” presentation).
Effector Mechanisms of Rejection and Graft-versus-Host Disease
While cytotoxic T cells are important effectors of graft rejection and GVHD, additional mechanisms involve effector cells of the innate immune system and cytokines as final mediators of tissue destruction. The net result of this multiplicity of pathways is considerable redundancy of mechanisms of graft rejection and GVHD.
Cytotoxic Mechanisms of Graft Rejection and Graftversus-Host Disease. Rejecting organs contain proteins and messenger ribonucleic acid (RNA) encoding perforin, granzymes, and proteases associated with cell-mediated cytotoxicity.
290,291,292,293,294,295,296,297 The presence in urine of RNA encoding perforin and granzyme B has been associated with renal allograft rejection in humans.
298 Although the perforin/granzyme pathway is the major cytolytic pathway for CD8 T cells and CD4 cells tend to utilize the Fas/FasL pathway,
299 both subsets are capable of both types of cytolytic activity,
300,301 and the perforin pathway is available to both T-cell subsets mediating GVHD.
302 All of these cytotoxic proteins play contributory roles, and no single protein has been found to be critical for solid organ graft rejection,
303,304,305,306 GVHD,
307,308,309,310 or bone marrow graft rejection
311 in the presence of clinically relevant mismatches. Critical cytotoxic interactions have been identified in less relevant animal models involving Fas-dependent GVHD directed at isolated class II MHC disparities
310 and perforin-dependent rejection of K
b mutant class I-only mismatched heart allografts.
303 Fas ligand promotes lymphoid hypoplasia
312 and skin and liver GVHD,
312 and both Fas ligand and TRAIL are required for GVHD-related thymic destruction.
313 While the perforin-granzyme pathway contributes to GVHD,
310,312 the Fas pathway appears to be of greater overall importance. In contrast, the perforin/granzyme and TRAIL pathways predominate in antileukemic effects, especially of CD8 cells, and selective blockade of the Fas/FasL pathway may ameliorate CD8-mediated GVHD without eliminating GVL effects.
309,314,315,316,317,318
Non-Cytotoxic T-Lymphocyte Effector Mechanisms in Graft Rejection and Graft-versus-Host Disease. T cells can effect rejection of grafts whose parenchymal cells do not express the TCR ligand, indicating the existence of “indirect” effector mechanisms. Entire skin grafts can be rejected when only the APCs are foreign,
319 indicating that nonselective destruction of grafted tissue can occur. Several studies have implicated indirect CD4 cell-mediated rejection of skin
320,321 and cardiac
322 allografts. Replacement of graft endothelium by the host was shown to be needed for rejection through this indirect effector mechanism.
322 GVHD of the liver and intestine can be induced by donor T cells in MHC-deficient hosts receiving wild-type host DCs, suggesting that indirect effector mechanisms may also mediate tissue injury of GVHD.
323,324 However, CD4-mediated GVHD against MiHAs is markedly attenuated when the target antigens are expressed only on hematopoietic cells.
325 Thus, “indirect” effector mechanisms can destroy transplanted tissue or recipient tissue in the case of GVHD, but less efficiently than direct cytotoxic mechanisms.
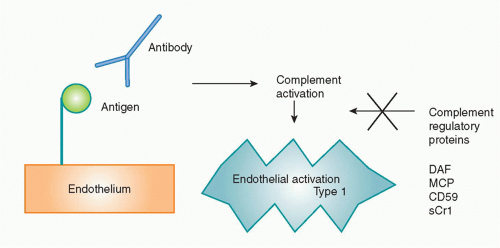
Another non-CTL graft rejection mechanism involves antibodies, which cause hyperacute rejection, acute humoral rejection, or chronic rejection through Fc receptor, complement-mediated, and other inflammatory pathways. B cells
326,327 and antibodies
327 contribute to cGVHD in animal models and are implicated in human cGVHD.
328,329,330 B-cell depletion with rituximab has been reported to have efficacy against cGVHD.
330,331,332Cytokines as Mediators of Graft Rejection and Graftversus-Host Disease. Interactions between alloreactive CD4 helper cells producing cytokines of the “Th1” type and alloreactive cytotoxic CD8+ effector cells can mediate rejection and GVHD via direct cytotoxicity.
333 The “indirect” mechanisms of graft rejection and GVHD are likely to include cytokines.
298 “Th17” cells producing IL-17 and other proinflammatory cytokines promote rejection and GVHD.
334,335,336,337,338,339,340,341,342,343,344,345,346,347,348 The generation of Th-17 cells is antagonized by Th-1 cells and promoted by IL-23, transforming growth factor (TGF)-β, and IL-6. A great redundancy of rejection pathways is suggested by studies detecting both Th1 (IL-2, IFNγ) and Th2 (IL-4, IL-5, IL-10) cytokines in rejecting allografts.
296,349,350,351,352,353,354,355,356 Th2 can also mediate GVHD and rejection. There appears to be strain-dependent tissue specificity to the type of GVHD induced by the various Th subsets.
337,357,358 Thus, while the concept that Th2 cytokines are anti-inflammatory attracted interest in the transplantation field,
359,360,361,362,363,364,365,366,367 Th2 responses can clearly contribute to both graft rejection
359,368,369,370 and GVHD.
357,371,372,373 With a few special exceptions,
374,375,376,377 studies using various cytokine knockout mice as recipients have failed to reveal any single molecule that is essential for rejection
378,379,380,381,382,383,384 or GVHD.
371,385,386
In GVHD, cytokines such as tumor necrosis factor (TNF)-α and IFNγ play a role. Macrophages are activated by lipopolysaccharides from the damaged gut epithelium and by IFNγ to release TNF-α, nitric oxide, and other mediators of tissue injury.
387,388,389,390,391 In certain models, TNF-α had been shown to be critical for wasting disease and intestinal GVHD.
392,393 While the relative contribution of cytokine-dependent mechanisms versus direct cell-mediated cytotoxicity to GVHD is still a matter of debate, GVHD is induced by T cells incapable of both perforin-mediated and Fas-mediated cytotoxicity, even in recipients lacking TNF receptor 1-mediated signaling,
310,394,395 demonstrating the redundancy of GVHD effector mechanisms.
Graft-Infiltrating Cells. Many types of cells infiltrate rejecting grafts, including CD4+ and CD8+ T cells, NK cells, and macrophages.
396,397,398,399,400,401,402,403,404 While B cells may be less prominent,
405 their presence has been associated with both acute and chronic rejection, and they are attracting increasing interest for their role not only as producers of antibody effectors of rejection, but also as APCs.
406 B cells may be located in tertiary lymphoid organs found in chronically rejecting allografts.
407
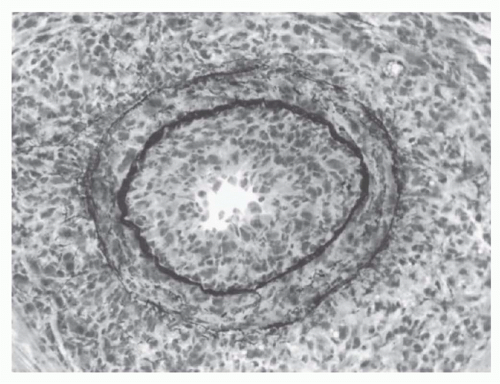
The number of invading T cells in a graft is not necessarily correlated with the speed of rejection.
405 This finding has
suggested that certain critical elements of the graft, such as its blood vessels, are the actual site of graft destruction and, indeed, endothelialitis is an important hallmark of clinically significant rejection activity.
408Repertoire analysis of graft-infiltrating T cells in acutely rejecting grafts reveals marked polyconality,
22,409,410,411,412 but only the donor-reactive CTLs show evidence of having been activated in vivo.
413 Oligoclonal dominance has been suggested in studies of tolerated rodent allografts
412 and in long-term rejected human kidneys.
414 T cells infiltrating xenografts included a broad TCR repertoire.
415,416,417 T cells mediating GVHD in the setting of multiple minor histoincompatibilities demonstrated a markedly skewed repertoire involving several different Vβ families.
418,419 Clinical studies suggest that the anti-MiHA TCR repertoire is most often polyclonal.
420Role of Natural Killer Cells. Although the role of NK cells in marrow rejection is well established in mice, the amount of resistance mediated by NK cells to allogeneic hematopoietic stem cells is limited and can be readily overcome by increasing the dose of donor stem cells administered.
421,422 Furthermore, a role for NK cells in resisting human allogeneic marrow engraftment has not been clearly demonstrated, although they might be expected to be important in recipients of reduced toxicity conditioning regimens. Indeed, patients with severe combined (T- and B-cell) immunodeficiency who have functional NK cells require cytotoxic conditioning to permit engraftment of haploidentical marrow, whereas those lacking NK cells do not.
423
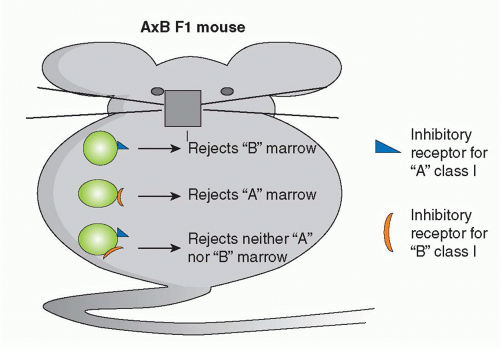
The ability of NK cells to be triggered by “missing self” may have utility in HCT.
424 Donor-derived NK cells with graft-versus-host reactivity due to the lack of donor class I MHC inhibitory ligands in the recipient can kill residual host leukemia cells and alloreactive cells that resist the marrow graft without causing GVHD. The alloreactive donor NK cells may also reduce susceptibility to GVHD by killing recipient APCs needed to activate donor T cells.
424,425 While striking antileukemic effects of KIR mismatching were detected in heavily conditioned patients receiving high doses of haploidentical CD34+ stem cells,
424 the effect of KIR incompatibility has been more variable in other clinical studies,
426,427,428,429,430,431 and the antitumor benefit is most evident for acute myelogenous leukemia.
432The possible role of NK cells in rejecting solid organ grafts is somewhat controversial. NK cells are prominent among cells infiltrating rejecting organ allografts and may be the earliest producers of inflammatory cytokines and chemokines and inducers of DC maturation.
433,434,435,436 If NK cells make an important contribution to solid organ allograft rejection under normal circumstances, they must be dependent on T cells, as mice lacking T cells are unable to reject nonhematopoietic allografts. Furthermore, whereas bone marrow allografts from class I deficient donors (β2m-/-) are subject to potent NK-mediated rejection (because these cells cannot trigger inhibitory receptors on host NK cells
437), β2m-/- skin grafts are not rejected by β2m+ recipients.
438 NK cells have recently been reported to play a critical role in cardiac allograft rejection in CD28 knockout mice,
439,440 and NK cells can mediate a particular form of chronic allograft vasculopathy in a murine cardiac allograft model.
435 This lesion may be triggered by viral infection.
441Inhibitory receptors on NK cells are quite broad in their class I specificity,
442 and fully allogeneic class I MHC marrow is less susceptible to NK-mediated marrow destruction compared to class I-deficient marrow.
437,443 Because of the increased disparity of xenogeneic compared to allogeneic MHC molecules, NK cells may receive fewer inhibitory signals from xenogeneic than allogeneic cells. Indeed, transduction of HLA molecules into porcine endothelial cells reduces NK cell-mediated xenogeneic cell adhesion and cytotoxicity.
444,445,446 However, some inhibitory receptors, such as killer cell lectin-like receptor G1, do recognize xenogeneic ligands such as e-cadherin.
447 NK cells may also be activated by interactions of activating receptors with ligands on xenogeneic cells,
448,449 of which several examples have been identified.
450,451 On balance, activating xenogeneic NK cell-target interactions are more effective than inhibitory interactions. Indeed, NK cells resist xenogeneic marrow engraftment to a greater extent than allogeneic marrow.
421,452,453,454 NK cells have also been implicated in the acute vascular rejection
455 that can destroy solid organ xenografts that have escaped hyperacute rejection (see following discussion) and in xenogeneic skin graft rejection.
456 As one mechanism by which NK cells mediate cytolysis is antibody-dependent cell-mediated cytotoxicity, it is possible that immunoglobulin G natural antibodies play a significant role in initiating NK cell-mediated rejection. NK cells also release cytokines, such as IFNγ, and TNF-α, which activate macrophages and endothelial cells, and induce inflammation.
455Role of Natural Killer T Cells. While NKT cells have apparent inhibitory effects on graft rejection
457,458,459,460 and GVHD,
461,462 they have also been reported to participate in rejection of tissues and bone marrow in mice.
463,464,465 The latter is due to the ability of NKT cells to activate NK cells.
465 NKT cells promote skin graft rejection by cross-primed CD8 cells via their ability to produce IFNγ.
466
Role of Monocytes/Macrophages and Eosinophils as Effectors of Rejection. Classical delayed-type hypersensitivity (DTH) responses are thought to depend on the activation of macrophages by helper T cells through production of IFNγ. It is likely that proinflammatory cytokines and chemokines produced by activated monocytes and macrophages play a role in endothelial cell activation and lymphocyte recruitment. Additionally, activated macrophages may damage tissue through the production of toxic molecules such as nitric oxide.
467
Macrophages play an especially important role in the rejection of cellular xenografts such as islets
468,469 in a T cell-dependent manner.
470 Macrophages cause almost immediate rejection of xenogeneic bone marrow, even in the absence of adaptive immunity.
471,472,473 Human macrophages can phagocytose porcine cells in an antibody- and complementindependent manner.
474 Additional studies have implicated macrophages in solid organ and skin xenograft rejection.
475,476,477,478,479,480 This prominent role for xenogeneic macrophages may reflect the combined ability of certain xenogeneic
receptors to activate macrophages,
481 whereas important inhibitory interactions, such as that between CD47 and its macrophage ligand SIRPα, are not effective.
482 Surprisingly, a system for monocyte-mediated recognition of allogeneic non-MHC nonself has been described.
483Eosinophils recruited to allografts by Th2 T-cell responses have been reported to be effectors of graft rejection in some experimental models, and eosinophils are often found clinically in rejecting allografts.
333 Th2-derived IL-4 and IL-5 recruit and activate eosinophils, which release highly cytotoxic substances from granules into the tissue.