Osteogenesis imperfecta symptoms
Atypical fractures
Short stature
Scoliosis
Blue sclera
Hearing loss
Dentinogenesis imperfecta
Laxity of the ligaments
Wormian bones
Laxity of the skin
The types of OI can be divided according to their severity (Table 26.2):
Table 26.2
Clinical characteristics according to the type of OI
Types of OI | Severity of fractures | Stature | Sclera | Hearing loss | Dentinogenesis imperfecta |
|---|---|---|---|---|---|
I | Slight (<100) | Normal–slightly lower | Blue | Present in 50 % | Rarely |
II | Perinatal death—multiple | Severely low stature | Blue | – | Yes |
III | Severe—multiple | Very low | Blue at birth | Frequent | Yes |
IV | Mild to moderate | Variable | Normal | Sometimes | Sometimes |
V | Moderate—multiple | Variable | Normal | No | No |
VI | Moderate | Slightly lower | Normal—discretely blue | No | No |
VII | Moderate | Slightly lower | Normal—discretely blue | No | No |
VIII | Severe/lethal | Short members dwarfism | Normal | Not related | No |
IX | Severe/lethal | Short limbs | Blue | No | Yes |
Mild (type I): Patients with type I OI suffer from few or no fractures before puberty, or sometimes numerous fractures throughout their lives. The deformities are minimal, and the individuals usually have lower than normal stature. Frequently, no fractures occur before the child begins to walk. The long bones of the arms, legs, and ribs are most often affected, as well as the small bones of the hands and feet. The frequency of fractures decreases after puberty. In adults there is premature osteoporosis and early hearing loss.
Moderate to severe (type III–IX): These children have a high frequency of fractures, moderate-to-severe bone deformities, kyphoscoliosis, short stature, and progressive hearing loss, with some children unable to move around. Adults exhibit early osteoporosis and hearing loss, more severe when compared with the mild form. Pregnant women exhibit accelerated bone loss during pregnancy and breastfeeding. Hypermobility of the joints can lead to pain and diminished function.
Lethal form (type II): Patients with the lethal perinatal form usually have intrauterine death, or die during early childhood. Fractures and respiratory insufficiency are frequent causes of death. In such cases, genetic counseling should be provided for families affected.
Diagnosis
A diagnosis of OI should be considered in any child with recurrent fractures from minimal trauma [3]. Family history, clinical examination, and radiological findings are important for diagnostic confirmation. The clinical picture is not always characteristic. Extra-skeletal manifestations may be subclinical (hearing loss), nonspecific, or more common at some ages (dentinogenesis imperfecta is most notable in the first dentition).
Imaging studies: X-rays of long bones and the spine may show fractures, bone calluses, or deformities, and the X-rays of the skull may reveal the presence of wormian bones.
Laboratory: Assessment of calcium metabolism (serum calcium, phosphorus, alkaline phosphatase, and PTH) is useful to rule out any preexisting hypocalcaemia or hyperparathyroidism. In cases involving OI, the parameters are usually normal. In type VI OI, increases in alkaline phosphatase may occur. Hypercalciuria is common in children with OI, and the magnitude of the urinary calcium loss reflects the severity of bone disease (shorter height, and higher rate of fracture). Bone formation markers are usually low and markers for bone resorption (CTX) usually high, especially in the more severely affected patient [10, 11].
Differential Diagnosis
Child victims of trauma, as well as patients with severe OI, may exhibit multiple fractures at different stages of fusion. In a study involving 39 children (older than 1 year of age) with fractured collarbones, 82 % were considered to be the result of child abuse, 8 % accidental injury, and 8 % bone fragility, and in one case fractures were the result of osteogenesis imperfect itself [12].
Another study evaluated 61 children victims of child abuse. After reviewing the medical records, 33 cases were confirmed as OI. The median age at examination was 7.1 months. All patients had fractures, 14 exhibited pain, 7 swelling, 5 showed limited movements, and 2 showed an abnormal position of their limbs. Radiographic findings consistent with OI were found in 19 of 33 patients (58 %); clinical findings were present in 23 of 33 patients (70 %), and a family history of OI in 55 % [13]. Therefore, in cases where child abuse is suspected, OI should always be considered, since any error in diagnosis can lead to serious consequences for the children and their families.
The clinical picture of OI is not always characteristic. Patients with mild OI exhibit no fractures until they start to move around. Retinal hemorrhage, subdural hematoma, and bruising may also occur as indirect signs of trauma.
Rickets may cause slow growth, bone deformities, elevated alkaline phosphatase, defects in bone mineralization, and, in some cases, abnormal formation of the teeth. Abnormalities in the sclera and hearing loss typically do not occur and on X-rays, epiphyseal plate enlargement is present. In the adult patient, osteomalacia may cause bone pain, fractures, and elevated alkaline phosphatase, also without causing hearing loss or blue sclera. Radiological findings include decreased bone density, pseudofractures, and loss of trabecular definition.
Other rare skeletal syndromes causing bone fragility and deformities should also be considered in the differential diagnosis of OI and these include Bruck syndrome, osteoporosis pseudo-glioma syndrome, polyostotic fibrous dysplasia, and juvenile Paget’s disease, hypophosphatasia, and idiopathic juvenile osteoporosis.
Treatment
The focus of treatment should be multidisciplinary in order to oversee early care and minimize complications. The objective is to reduce the rate of fractures, prevent deformities, decrease chronic pain, and improve functional capacity.
Bisphosphonates are the main therapeutic agents used to prevent fractures in most forms (except for type VI) although none have been approved specifically for use in children and adults with OI. Bisphosphonates are stable pyrophosphate analogues, and are potent inhibitors of bone reabsorption and bone turnover.
The majority of studies was conducted in children and did not include control groups. Observed benefits included an increase in bone mineral density, reduced fracture rate, and improvement in mobility and pain [14]. Pamidronate is administered intravenously during consecutive 3-day cycles, with 2–4-month intervals, using 0.5 mg/kg/day up to 1 mg/kg/day depending on the age (Table 26.3).
Table 26.3
Administration of pamidronate in children with OI
Age | Dose of pamidronate | Frequency |
|---|---|---|
<2 years | 0.5 mg/kg/day for 3 days | 2/2 months |
Between 2 and 3 years | 0.75 mg/kg/day for 3 days | 3/3 months |
>3 years | 1.0 mg/kg/day for 3 days | 4/4 months |
Short-term adverse effects on bone quality and fracture healing are not present, despite the significant reduction in bone turnover with bisphosphonate treatment [15]. Linear growth does not appear to be affected and the greatest benefit seems to occur within the first 2–4 years of therapy. It is prudent to reserve pamidronate for patients in whom the clinical benefits outweigh the risks (deformity of long bones, vertebral compression fractures, and three or more fractures per year) since the long-term effects as yet are not well understood [16].
In a meta-analysis, oral risedronate (35 mg per week) for 24 weeks led to bone mineral density increases by 3.9 % at the lumbar spine without statistical significance in the total hip measurements. Bone pain did not improve significantly, and the fracture rate remained high [17].
In most cases, orthopedic care focuses on fractures, but also should be considered for the correction and prevention of deformities, especially in the lower limbs, including surgical treatment.
Osteomalacia
Osteomalacia is not a common metabolic bone disease, but is often neglected especially in its early stages because of the nonspecific nature of symptoms such as vague bone pain and muscle weakness [18]. The disease is characterized by a generalized weakening of bone, leading to deformity, and it is often caused by defects occurring at any step of the metabolism or action of vitamin D [19]. It occurs mostly due to dietary deficiency and exposure to sunlight, but can also be due to intestinal malabsorption, chronic kidney failure, or vitamin D resistance [18] (Table 26.4). Osteomalacia can also occur in patients with primary hypophosphatasia (Table 26.5) due to one of the syndromes of hereditary hypophosphatemia (X-linked, autosomal dominant and with hypercalciuria) or oncogenic due to fibroblastic growth factor-23 (FGF-23) secretion by the tumor [20, 21].
Table 26.4
Etiology of osteomalacia
Causes of osteomalacia |
|---|
Vitamin D deficiency |
Lack of sunlight |
Malabsorption syndrome |
Liver diseases |
Chronic renal insufficiency |
Anticonvulsants |
Reduced calcium intake |
Heavy metals: Aluminum, lead, cadmium |
Table 26.5
Etiology of hypophosphatemic osteomalacia
Causes of hypophosphatemic osteomalacia |
|---|
X-linked |
Autosomal dominant |
With hypercalciuria |
Oncogenic osteomalacia |
There is an increasing prevalence of vitamin D deficiency in many countries, even in those close to the equator, and those with abundant sunlight [22]. The at-risk population includes the elderly with little exposure to sunlight as well as patients with poor absorption including those with celiac disease and those submitted to gastrointestinal bypass surgery. Likewise, individuals living in cold weather climates, and women who wear clothes that cover almost the entire body area, are also predisposed to hypovitaminosis D [19].
The main histological findings are an excessive accumulation of bone matrix that is not, or only poorly, mineralized, decreased bone volume, increased accumulation of osteoid, and increases in the osteoid thickness of bones and the surface area [23].
Clinical Manifestations
Osteomalacia may be asymptomatic. When symptomatic, general symptoms include chronic bone and muscle pain, weakness, fatigue, difficulty in walking, and a high risk of fractures due to bone fragility. Deformities related to the softening of the adult skeleton include kyphosis, pectus carinatum, a decrease in stature, genu varum, and acetabular protrusion [23].
Bone pain seems to be caused by hydration of the demineralized bone matrix beneath the periosteum, which is extended, causing compression of nerve terminals. It is usually persistent, diffuse, and symmetrical, starting in the lower back and spreading to the pelvic girdle, hip, and ribs. Pain on palpation of these sites is an important clinical sign. Muscle weakness is usually proximal and associated with hypotonia, atrophy, and discomfort during movement [18].
Diagnosis
The most characteristic laboratory findings are a lower serum calcium level, a decrease in urinary calcium levels, hypophosphatemia, and increased levels of alkaline phosphatase (ALP) (Table 26.6). Increased ALP activity is the most frequent and earliest marker for osteomalacia, and reflects the activity of the osteoblast, which forms the demineralized matrix [24]. The key test for the diagnosis of vitamin D deficiency is the demonstration of decreases in serum 26OHD [25].
Table 26.6
Laboratory findings in nutritional osteomalacia
Laboratory results for osteomalacia | |
|---|---|
Elevated PTH | 100 % |
25 (OH) Vitamin D < 15 ng/ml | 100 % |
Elevated alkaline phosphatase | 95 % |
Low urinary calcium | 87 % |
Low calcium and phosphate levels | 27 % |
PTH may be typically increased. As vitamin D deficiency increases, the hypersecretion of PTH leads to bone remodeling and endocortical bone reabsorption, resulting in cortical bone loss and increased risk of fractures. In addition, biochemical markers for bone turnover may be increased [18].
On X-rays, the most characteristic feature is the Looser’s zone, a band adjacent to the periosteum which represents a stress fracture (cracks without repetitive posttraumatic displacement). It occurs most commonly in the ribs, pubis, and scapula (Fig. 26.1). This is in contrast to the fissures that occur in the long bones in Paget’s disease [25]. Bone scintigraphy usually shows areas of focal increases in MDP uptake (Fig. 26.2). Bone mineral density is usually decreased, mainly in the cortical [18].
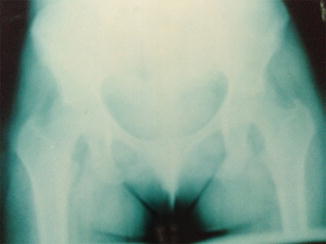
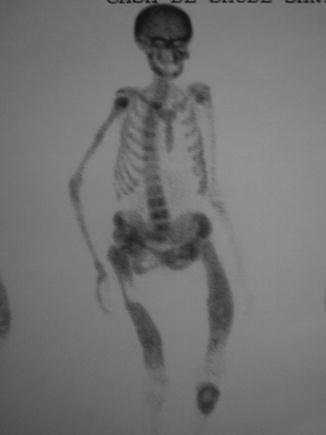
Fig. 26.1
Looser’s zone at the right pubis in a 32-year-old woman with hypophosphatemic osteomalacia
Fig. 26.2
Bone scintigraphy in a 52-year-old woman with primary hyperparathyroidism and osteomalacia. Serum 25OHD: 8 ng/ml
Although the diagnosis of osteomalacia can be carried out on the basis of clinical and laboratory findings, transiliac bone biopsy, with tetracycline labeling, can help make a definitive diagnosis. This takes less than 30 min, under local anesthesia, with minimal discomfort to the patient. The specimen should be stored in 70 % alcohol and sent to a specialized laboratory for histomorphometric studies. The characteristic finding is impaired mineralization with an absent two-band tetracycline label [26].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


